1. Introduction
Ranitidine is a pharmaceutical pollutant found ubiquitously in aquatic environments due to its extensive use in treating peptic ulcers and gastroesophageal reflux diseases, as well as its ineffective degradation by conventional biological treatment processes [1–3]. High concentrations of ranitidine were detected in effluents from sewage treatment plants (dozens to hundreds of ng/L) and in surface water (dozens of ng/L) [4, 5]. Moreover, ranitidine exhibits a significant potential risk of generating the carcinogenic N-nitrosodimethylamine (NDMA) during the chloramine disinfection process [6–8]. 0.7 ng/L of NDMA in drinking water can elevate the lifetime cancer risk by one million times [9]. Therefore, there is an urgent demand for developing a practical method for degrading ranitidine as a precursor of NDMA.
Various techniques based on advanced oxidation processes (AOPs) have been developed and used for the degradation of ranitidine. They include energy-induced (UV-C and ultrasound [10]), photocatalytic (TiO2/UV light [11], MgIn2S4/g-C3N4/visible light [12], and MoS2/reduced graphene oxide/visible light [13]), and Fenton-like methods (nZVI/Ti3C2/H2O2 [14], zeolite impregnated with iron/H2O2 [15], and CoFe2O4/g-C3N4/peroxymonosulfate [16]). Although all methods previously reported have successfully degraded ranitidine, developing a more economical method that does not require expensive materials and high energy remains a challenging issue.
It has recently been proposed that the catalytic activation of H2O2 using nonferrous metal oxides, such as TiO2 and WO3, is an economical Fenton-like system for oxidizing arsenite to arsenate [17, 18]. The nonferrous metal oxide/H2O2 system operates under ambient conditions without the need for high-energy irradiation. The highly stable nature of nonferrous metal oxides allows for repeated use, contrasting with iron-based catalysts in Fenton-like systems [19]. Importantly, nonferrous metal oxides, especially TiO2, are cheap and non-toxic [20, 21].
Some chelating agents, such as oxalate, citrate, ethylene diamine tetraacetic acid, carboxy-methyl-β-cyclodextrin, tartrate, and succinate, have the ability to enhance the activity of Fenton-like systems using iron-based catalysts (i.e., ferrous ions (Fe2+) and magnetite (Fe3O4)) [22, 23]. The complexation of the aforementioned organic compounds with Fe2+ increases the reducing power of Fe2+ by shifting the reduction potential of FeII-organic complexes to low values [24, 25]. Consequently, the production of hydroxyl radicals (•OH) through H2O2 reduction and the degradation of phenolic pollutants were greatly amplified in the presence of these organic chelating agents.
In this study, we examined the effect of oxalate on the degradation of ranitidine in the TiO2/H2O2 system under dark conditions. Notably, the investigation into the effect of a chelating agent in the nonferrous metal oxide/H2O2 system remains unexplored. The degradation kinetics of ranitidine were measured in the TiO2/H2O2/oxalate system under various conditions, and the results were discussed. The degradation mechanism of ranitidine in the TiO2/H2O2/oxalate system was investigated through the analysis of reactive oxygen species (ROS) and ROS-quenching experiments. In addition, the effects of other chelating agents in the TiO2/H2O2 system and the effect of oxalate in the WO3/H2O2 system were examined.
2. Experimental Section
2.1. Materials and Reagents
The materials and reagents were used as received without additional purification; they encompass titanium dioxide (TiO2, Hombikat UV100 (Sachtleben Chemie GmbH) and Aeroxide P25 (Evonik)), tungsten(VI) oxide (WO3, Sigma-Aldrich), ranitidine hydrochloride (C13H22N4O3S·HCl, Sigma, ≥98.0%), cimetidine (C10H16N6S, 100%, Sigma), iron(II) perchlorate hydrate (Fe(ClO4)2· xH2O, ≥98.0%, Aldrich), hydrogen peroxide (H2O2, 30.0%, Daejung), sodium acetate (C2H3NaO2, 99.0%, Sigma-Aldrich), malonic acid (C3H4O4, ≥99.0%, Sigma-Aldrich), sodium citrate tribasic dihydrate (C6H5Na3O7·2H2O, ≥99.0%, Sigma-Aldrich), potassium oxalate monohydrate (C2K2O4·H2O, ≥99.0%, Fluka), Horseradish peroxidase (POD, type VI-A, Sigma-Aldrich), N,N-diethyl-p-phenylenediamine (DPD, C10H16N2, 97.0%, Aldrich), ascorbic acid (C6H8O6, 99.6%, Junsei), sodium azide (NaN3, ≥99.5%, Sigma-Aldrich), tetranitromethane (CN4O8, 100%, Aldrich), coumarin (C9H6O2, ≥99.0%, Sigma), phosphoric acid (H3PO4, 85.0%, Junsei), acetonitrile (CH3CN, ≥99.0%, J. T. Baker), perchloric acid (HClO4, 60%, Sigma-Aldrich), and sulfuric acid (H2SO4, ≥95.0%, Sigma-Aldrich). All suspensions and solutions were prepared using deionized water (resistivity > 18.0 MΩ·cm) produced by a water purification system (Millipore Milli-Q).
2.2. Experimental Procedure
Hombikat UV100 TiO2 powder was dispersed in deionized water through sonication for 2 min using an ultrasonic cleaner (SciLab). Aliquots of oxalate and ranitidine (or cimetidine) stock solutions were then introduced into the TiO2 suspension. Subsequently, the pH of the suspension was adjusted to 5.0 using an HClO4 solution. The suspension was stirred for 30 min before the addition of H2O2 to exclude the removal of ranitidine by adsorption from the total degradation. Finally, H2O2 was introduced into the TiO2 suspension containing oxalate and ranitidine. This moment was considered the start of the reaction. Generally, the initial concentrations of TiO2, H2O2, oxalate, and ranitidine were 15 mg/30 mL, 1 mM, 500 μM, and 100 μM, respectively.
The degradation reaction proceeded in a 50 mL beaker covered with aluminum foil, serving as the reactor, with continuous magnetic stirring under dark conditions. At specified time intervals, sample aliquots (1.5 mL) were extracted from the reactor and filtered through a 0.45 μm polytetrafluoroethylene filter (Millipore) to eliminate TiO2 particles. The filtered samples were promptly measured. All experiments were replicated two or three times to validate data reproducibility, and the figures represent average values with standard deviations.
2.3. Chemical Analyses
The concentration of ranitidine and cimetidine was assessed using high-performance liquid chromatography (HPLC, Agilent 1260 Infinity II) equipped with a Poroshell 120 EC-C18 column (4.6 mm × 150 mm) and a variable wavelength detector. The eluent comprised a binary mixture of acetonitrile and a 0.1% phosphoric acid solution, with a volume ratio of 5:95. The detection wavelength was set at 218 nm for cimetidine and 228 nm for ranitidine. The column temperature was maintained at 30 °C, and the eluent flowed at a rate of 1.0 mL/min. The concentration of total organic carbon (TOC) was determined using a TOC analyzer equipped with a nondispersive infrared sensor, serving as a CO2 detector (Shimadzu TOC-LCPH). The TOC concentration was calculated by subtracting the concentration of inorganic carbon from the total carbon concentration.
The concentration of H2O2 was determined using the colorimetric DPD method employing the DPD/POD reagent, which consists of 100 mg of DPD, 10 mg of POD, 10 mL of water, and 10 mL of H2SO4 (0.1 M) [26, 27]. Specifically, 3 mL of the 20-fold diluted sample was mixed with 0.4 mL of phosphate buffer solution (0.5 M) and 0.1 mL of DPD/POD reagent. The mixture in a conical tube was vigorously shaken for 1 min, and its absorbance was measured at 551 nm (ɛ of DPD•+ = 21,000 M−1 cm−1) using a UV–visible spectrophotometer (Agilent Cary 60). To assess the production of •OH, the fluorescence emission intensity of 7-hydroxycoumarin was measured at 460 nm using a spectrofluorometer (Horiba FluoroMax 4), with 332 nm UV light employed as the excitation source [28, 29]. This compound is formed through the reaction of •OH with coumarin (1 mM) (•OH + coumarin + O2 → 7-hydroxycoumarin + HO2•). For measuring the production of HO2•, the absorbance at 350 nm of nitroform (ɛ = 14,600 M−1 cm−1 at 350 nm), formed from the reaction between tetranitromethane (50 μM) and HO2• (HO2• + tetranitromethane → nitroform + NO2 + O2 + H+), was measured using a UV–visible spectrophotometer (Agilent Cary 60) [30, 31].
3. Results and Discussion
3.1. Effect of Oxalate on the Degradation of Ranitidine in the TiO2/H2O2 System
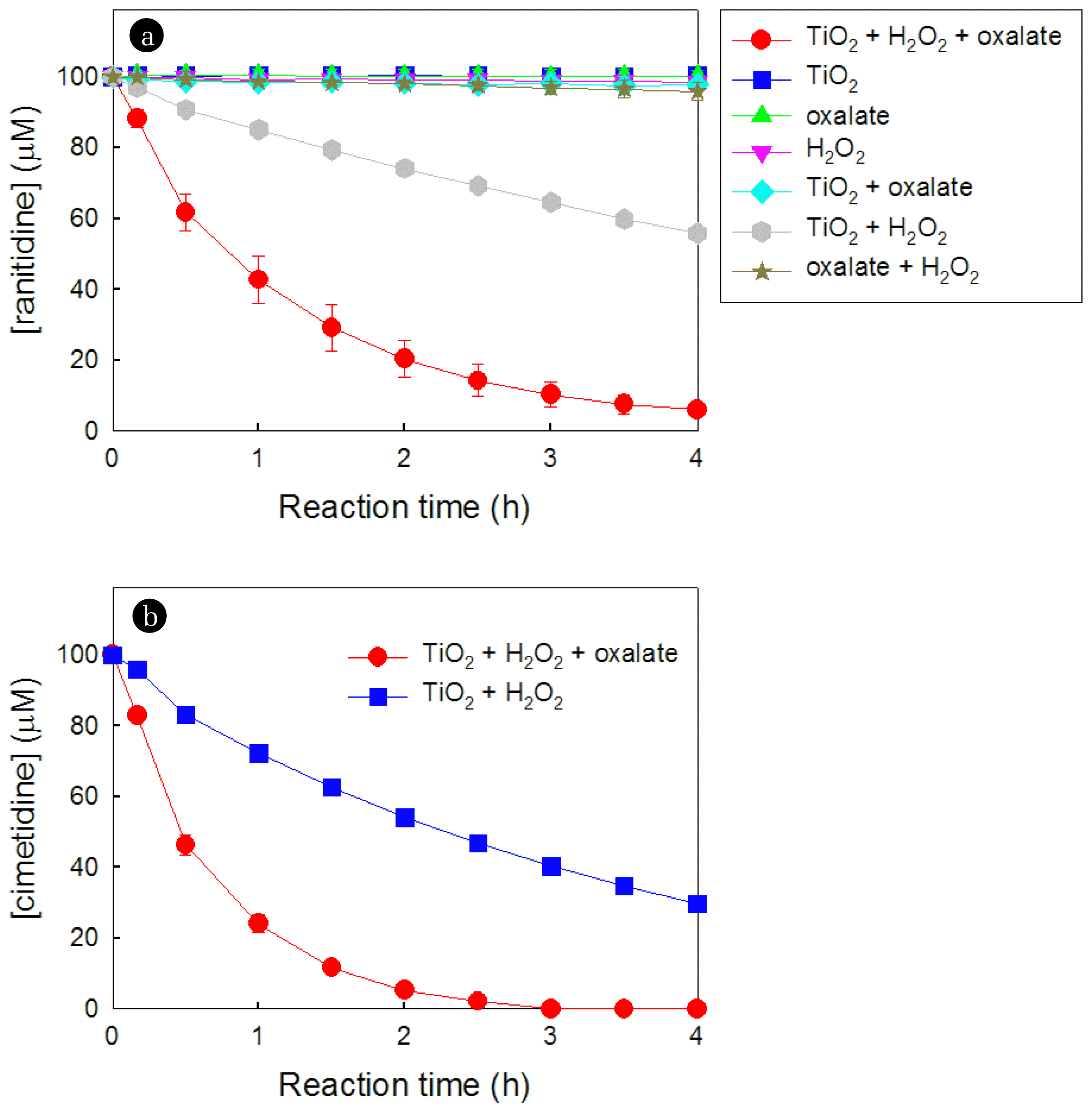
The effect of oxalate on the degradation of ranitidine in the presence of TiO2 and H2O2 was investigated and compared with control systems (Fig. 1a). The concentration of ranitidine diminished slowly in the TiO2/H2O2 system without oxalate. Remarkably, the introduction of oxalate to the TiO2/H2O2 system notably boosted the decomposition of ranitidine. After 4 h of reaction, 44.2% of ranitidine was degraded in the absence of oxalate, while the degradation reached 94.0% in the presence of oxalate. In terms of the pseudo-first-order degradation rate constant (k), the presence of oxalate resulted in a 5.7-fold increase in the degradation rate constant (i.e., 0.144 h−1 without oxalate → 0.823 h−1 with oxalate). In addition, the concentration of total organic carbon (TOC) in the TiO2/H2O2/oxalate system decreased from 27.4 ppm to 24.0 ppm after 4 h of reaction, resulting in a TOC removal efficiency of 12.4%. Other control systems (i.e., TiO2 alone, H2O2 alone, oxalate alone, TiO2 + oxalate, and oxalate + H2O2) did not induce any degradation of ranitidine. These results indicate that the presence of oxalate in the TiO2/H2O2 system could synergistically enhance the production of oxidant species, consequently resulting in the enhanced degradation of ranitidine.
We also tested the degradation of cimetidine, another model pharmaceutical pollutant, to verify the pervasiveness of the positive effect of oxalate in the TiO2/H2O2 system (Fig. 1b). After 3 h of reaction, cimetidine was completely degraded in the presence of oxalate, while 40.3 μM of cimetidine remained in the absence of oxalate. Similar to the case of ranitidine, the existence of oxalate markedly elevated the degradation rate constant of cimetidine from 0.304 to 1.470 h−1. This result implies that the positive effect of oxalate on the degradation process in the TiO2/H2O2 system is pervasive and is not restricted to a specific pollutant.
3.2. Detailed Mechanism of Oxidant Generation in the TiO2/H2O2/Oxalate System
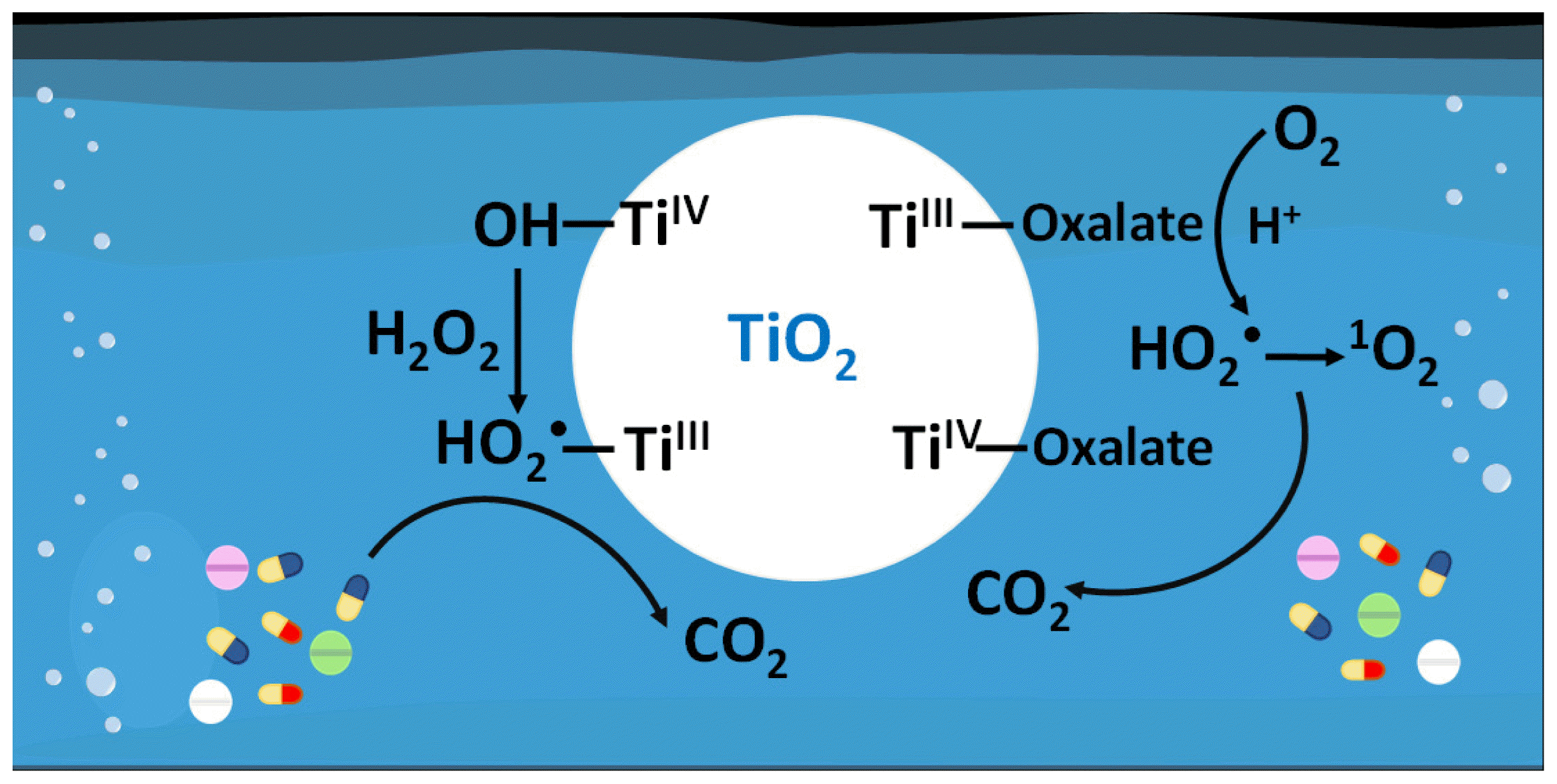
It has been reported that the catalytic decomposition of H2O2 on metal oxides can generate HO2• and •OH [17, 18]. The generation of HO2• is induced by the formation of peroxo surface complexes of H2O2 on metal oxides (–OOH groups) (Eq. (1)), followed by subsequent inner-sphere electron transfer from the –OOH groups to the metal centers (Eq. (2)) [32]. In addition, upon the adsorption of H2O2 on the metal oxide surface through hydrogen bonding, the metal center can induce the homolytic cleavage of the O–O bond in H2O2, resulting in the production of •OH (Eq. (3)) [33].
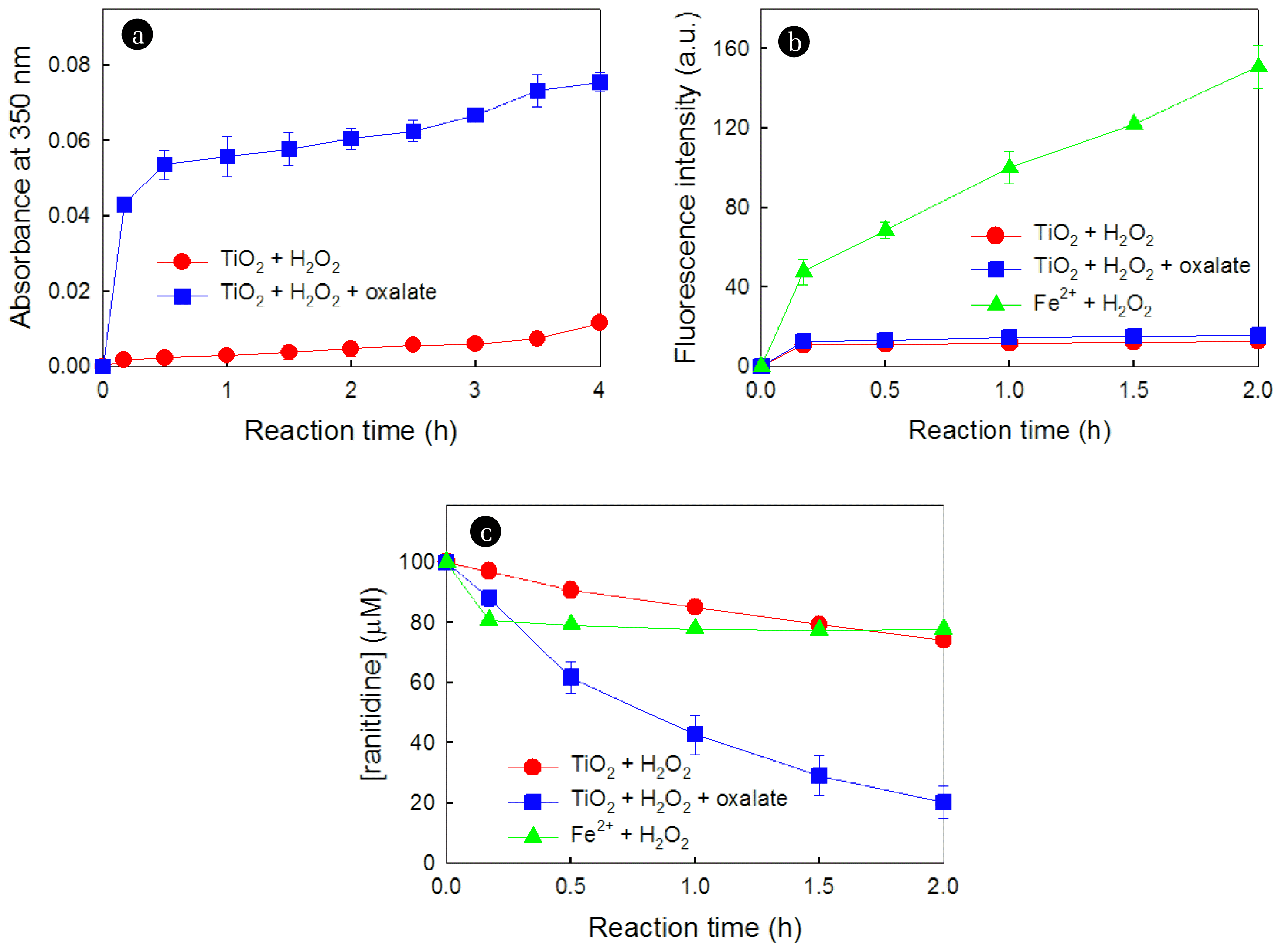
To establish the role of oxalate in the TiO2/H2O2 system, the production of radical species, such as HO2• and •OH, was measured and compared in the absence and presence of oxalate. The production of HO2•, directly associated with the formation of nitroform in the presence of tetranitromethane, was significantly accelerated in the presence of oxalate (Fig. 2a).
We also investigated the effect of oxalate in the TiO2/H2O2 system on the production of •OH using fluorescence spectroscopy, with coumarin employed as the •OH-trapping reagent. Although the production of •OH was observed in both the absence and presence of oxalate, the quantity was minimal, and the difference was not significant. In comparison to the Fenton reaction (Fe2+ + H2O2 → Fe3+ + OH− + •OH) [34], the production of •OH in the TiO2/H2O2 system, regardless of the presence of oxalate, was much smaller (Fig. 2b). In contrast to this behavior, the degradation of ranitidine was more significant in the TiO2/H2O2/oxalate system than in the Fe2+/H2O2 system (Fig. 2c). This result suggests that the involvement of •OH in the degradation of ranitidine within the TiO2/H2O2/oxalate system was marginal.
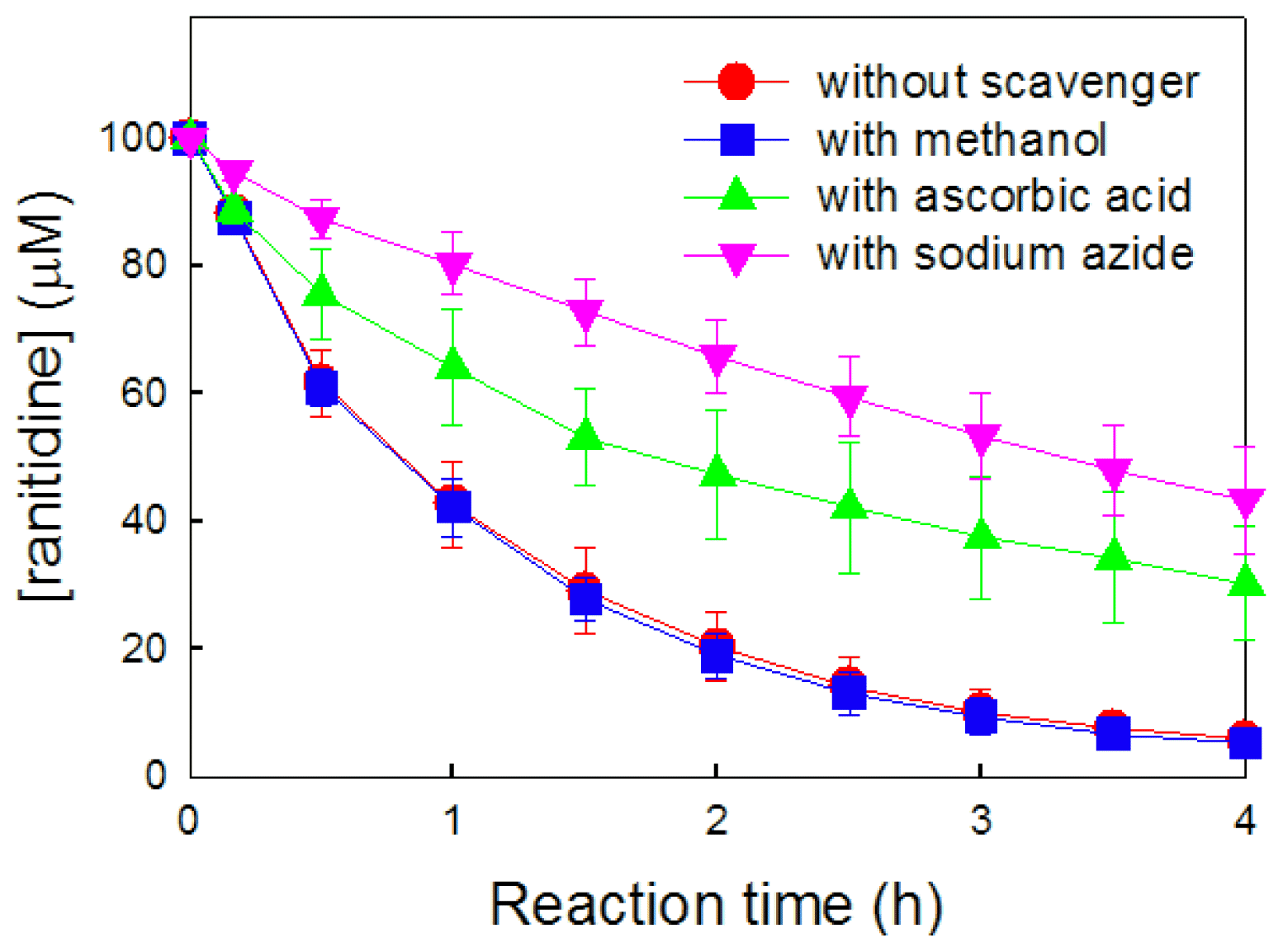
To confirm that HO2• acts as the primary oxidant for the degradation of ranitidine in the TiO2/H2O2/oxalate system, ROS-quenching experiments were conducted using various ROS scavengers, such as methanol for •OH (k = 9.7 × 108 M−1 s−1) [35], ascorbic acid for HO2• (k = 1.6 × 104 M−1 s−1) [36], and sodium azide for singlet oxygen (1O2) (k = 1 × 109 M−1 s−1) [37]. As illustrated in Fig. 3, methanol (•OH scavenger) exhibited an insignificant effect on the degradation of ranitidine, suggesting that •OH was not the primary oxidant in the TiO2/H2O2/oxalate system. This finding is in agreement with the marginal production of •OH in the TiO2/H2O2/oxalate system (Fig. 2b). In contrast, the addition of ascorbic acid (HO2• scavenger) reduced the degradation of ranitidine. This result implies that HO2• plays a significant role in the degradation of ranitidine in the TiO2/H2O2/oxalate system. In addition, when sodium azide, acting as a 1O2 scavenger, was present, the degradation of ranitidine was retarded. The reaction between HO2• and O2•− (also two O2•−) produces 1O2 as another oxidant for ranitidine degradation, as described by Eqs. (4)–(6) [38, 39]. Based on the results of ROS production and ROS-quenching experiments, both HO2• and 1O2 can be considered as the primary oxidants in the TiO2/H2O2/oxalate system. Although the oxidation power of •OH is higher than those of HO2• and 1O2 [40], weak oxidants, HO2• and 1O2, contribute more significantly to the degradation process in the TiO2/H2O2/oxalate system because the production of •OH through Eq. (3) is minimal (Fig. 2b).
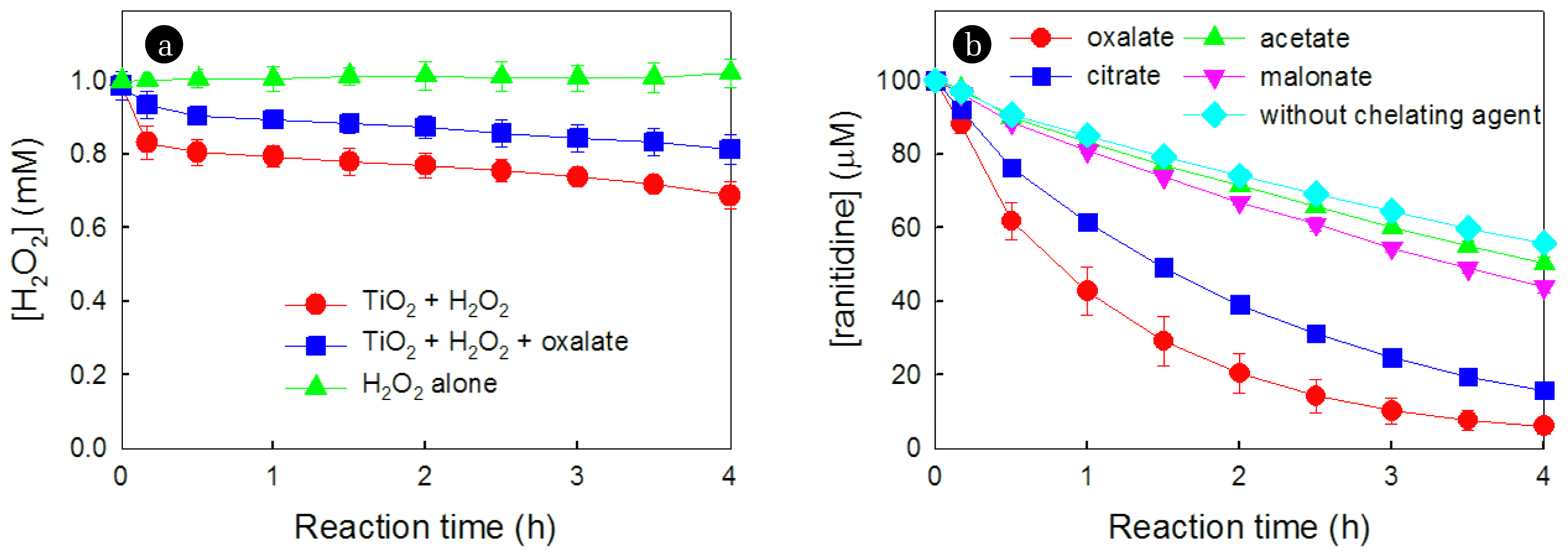
Fig. 4a depicts the time profiles of H2O2 decomposition induced by TiO2, both in the absence and presence of oxalate, over reaction time. Interestingly, the catalytic decomposition of H2O2 was lower in the presence of oxalate compared to its absence. The contrasting behaviors observed in the presence of oxalate, higher production of HO2• (Fig. 2a) but lower decomposition of H2O2 (Fig. 4a), suggest the existence of another pathway for the production of HO2• in the presence of oxalate.
Oxalate can form a complex with the TiO2 surface through monodentate and bidentate carboxylate linkages [41]. Due to the competition between H2O2 and oxalate for complex formation on the TiO2 surface, a lower decomposition of H2O2 was observed in the presence of oxalate (Fig. 4a). Although >TiIII (surface TiIII species on TiO2), which can act as a reductant, is generated through electron transfer from the –OOH groups to >TiIV (>TiIV–OOH → >TiIII–•O2H), the reduction of oxygen (i.e., the formation of HO2•) by >TiIII is not favored due to the more positive reduction potential of >TiIII (E0(Ti4+/Ti3+) = 0.1 VNHE) compared to the reduction potential of oxygen (E0(O2/HO2•) = −0.05 VNHE) [42, 43]. However, it has been reported that the complexation of oxalate with metal species decreases the reduction potential of metal species (i.e., increases the thermodynamic driving force for electron transfer) [22, 23]. For example, the reduction of oxamyl by Fe2+ was very slow, but it proceeded rapidly after complexation with oxalate (i.e., after the formation of FeII-oxalate complexes) [25]. Similarly, the complex between >TiIII and oxalate (i.e., >TiIII(oxalate)n(2n–3)−) would have a more negative reduction potential, facilitating the formation of HO2• through oxygen reduction (Eq. (7)).
To provide evidence for this hypothesis, the effects of various chelating agents in the TiO2/H2O2 system on the degradation of ranitidine were investigated (Fig. 4b). Although all chelating agents, such as acetate, malonate, citrate, and oxalate, enhanced the degradation of ranitidine, the extent of the positive effect varied significantly depending on the type of chelating agent. The positive effects of chelating agents followed the order: oxalate > citrate > malonate > acetate. This order exactly corresponds with the sequence of lower reduction potential values of FeII-organic complexes (oxalate < citrate < malonate < acetate) [25]. The observed behaviors support the hypothesis that the enhanced production of HO2• in the presence of oxalate is attributed to the negative shift in reduction potential resulting from the complexation between >TiIII and oxalate.
3.3. Investigation of Ranitidine Degradation Kinetics Across Diverse Conditions
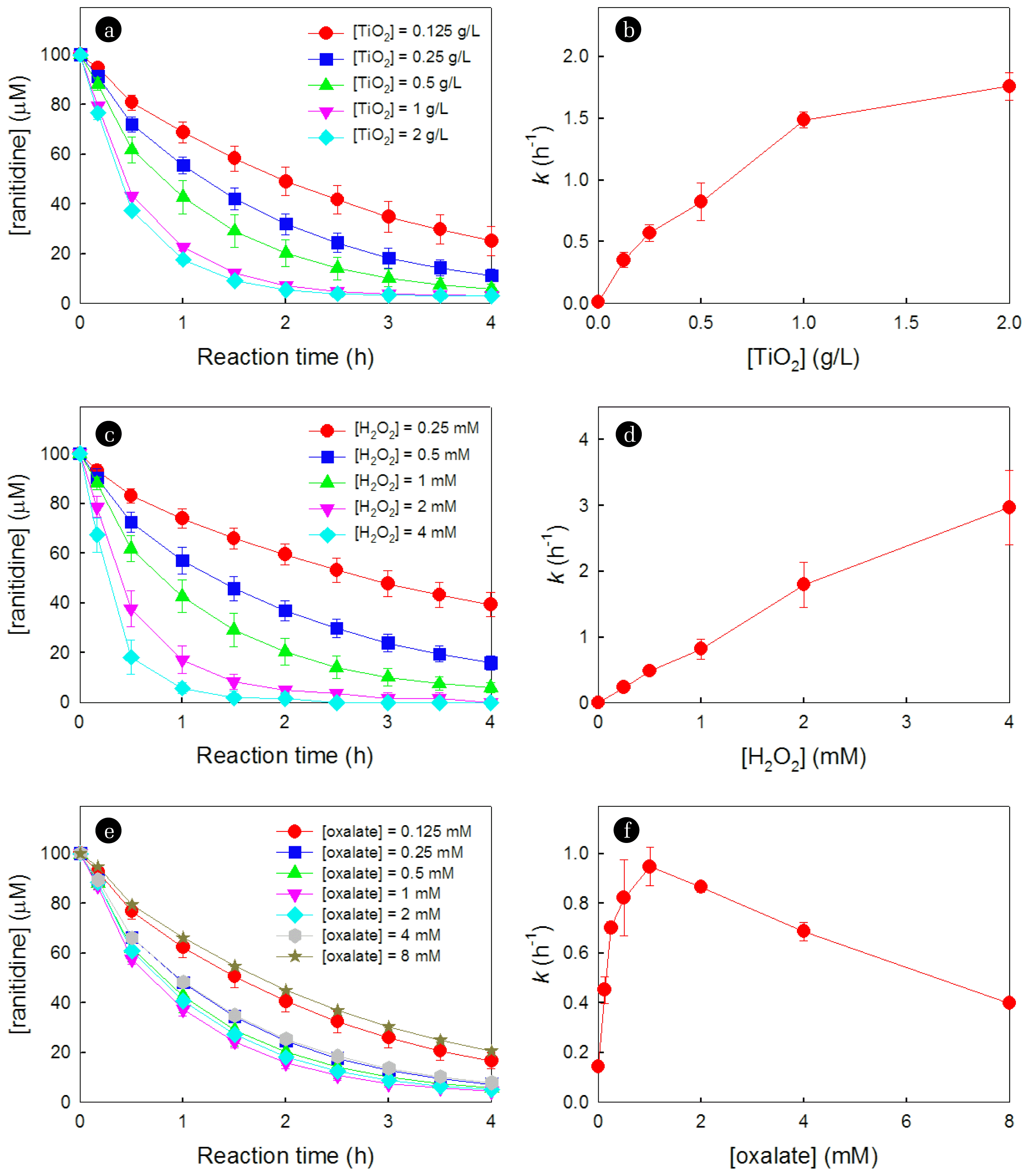
The degradation kinetics of ranitidine in the TiO2/H2O2/oxalate system were investigated as functions of TiO2 dosage, H2O2 concentration, and oxalate concentration (Fig. 5). The degradation rate (k) of ranitidine displayed a positive correlation with increasing TiO2 dosage until it reached saturation at [TiO2] = 1.0 g/L (Fig. 5a and b). A higher dosage of TiO2 provides more interaction sites among TiO2, H2O2, and oxalate, thereby enhancing the formation of TiO2-H2O2 complexes and TiO2-oxalate complexes. However, because the interaction sites on TiO2 were completely occupied by H2O2 and oxalate at [TiO2] = 1.0 g/L, a further increase in TiO2 dosage had a minimal impact on the degradation kinetics of ranitidine.
The k value increased linearly with increasing H2O2 concentration (Fig. 5c and d). Higher concentrations of H2O2, serving as an HO2• precursor and producing >TiIII as an oxygen reductant for HO2• generation, can increase the production of HO2• and 1O2, which can efficiently react with ranitidine. Therefore, the degradation was accelerated at higher concentrations of H2O2 in the TiO2/H2O2/oxalate system.
Contrary to the degradation patterns observed with changes in TiO2 dosage and H2O2 concentration, the k value increased drastically with increasing oxalate concentration up to 1 mM, but gradually decreased from 1 mM (Fig. 5e and f). Oxalate can lower the redox potential of >TiIII by forming a complex with TiO2, which facilitates the generation of HO2• through oxygen reduction. However, oxalate can compete with H2O2 for the formation of complexes on the TiO2 surface and act as a scavenger for HO2• and 1O2 [31]. The dual function of oxalate helps to explain the observed initial rise and subsequent decline in the k value as the oxalate concentration increases in the TiO2/H2O2/oxalate system.
3.4. Effect of Oxalate on the Degradation of Ranitidine in other Metal Oxide/H2O2 Systems
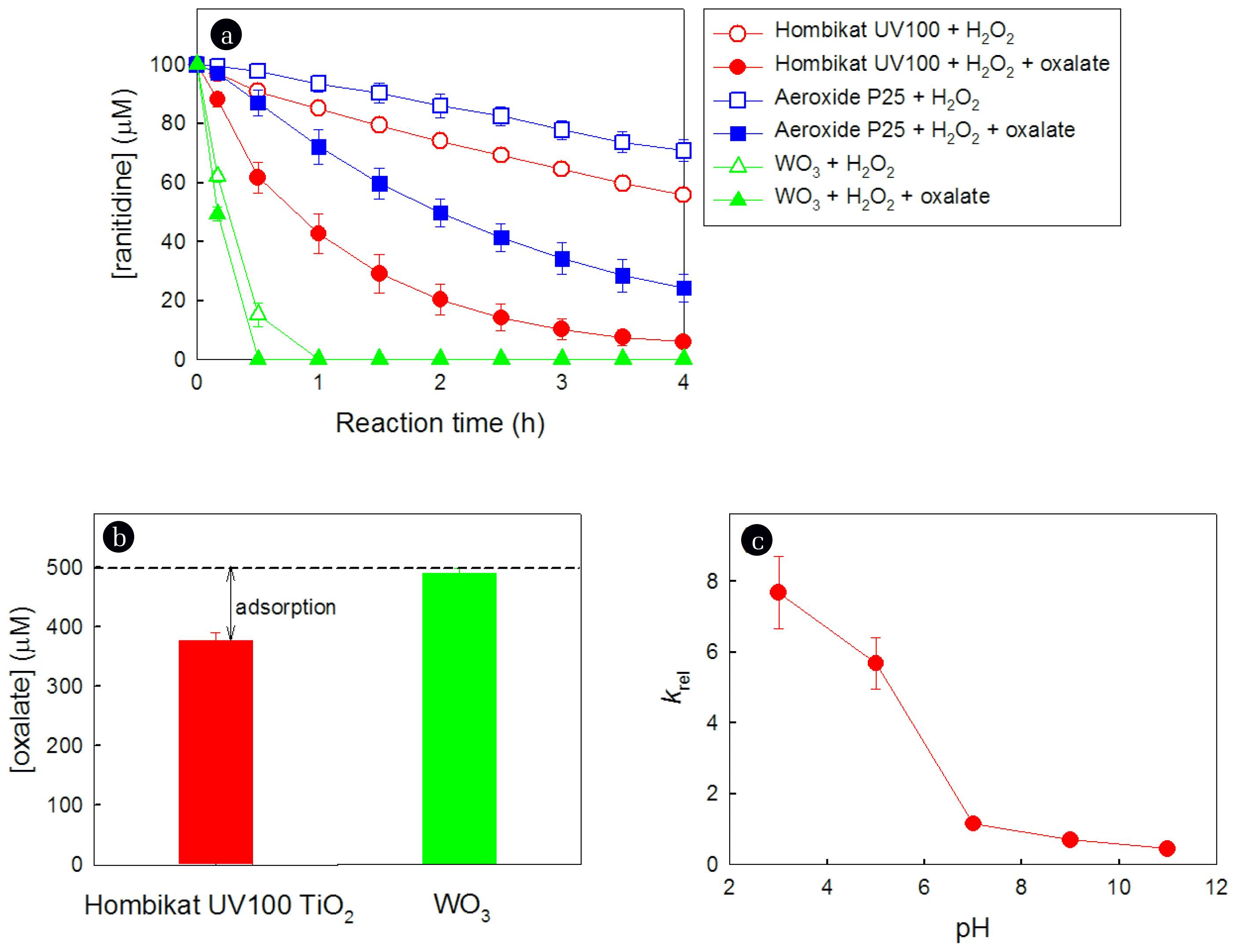
To determine whether the positive effect of oxalate is observed in other metal oxide/H2O2 systems, the effects of oxalate on the degradation of ranitidine were investigated in the Aeroxide P25 TiO2/H2O2 and WO3/H2O2 systems. These results were then compared with those obtained in the Hombikat UV100 TiO2/H2O2 system. Regardless of the type of metal oxide, the presence of oxalate enhanced the degradation of ranitidine (Fig. 6a). However, the extent of the positive effect of oxalate varied considerably depending on the type of metal oxide. The relative first-order degradation rate constants (krel), which represent the ratio of k with oxalate to k without oxalate, were 5.715, 4.114, and 1.443 in the cases of Hombikat UV100 TiO2, Aeroxide P25 TiO2, and WO3, respectively (Table 1). The positive effect of oxalate in the TiO2/H2O2 system was more pronounced than that in the WO3/H2O2 system.
The points of zero zeta potential (PZZP) of Hombikat UV100 TiO2 (pH 6.0) and Aeroxide P25 TiO2 (pH 6.3) are higher, whereas that of WO3 (pH 2.0) is lower than the pH of the catalyst suspension containing H2O2 and oxalate (5.0) [44, 45]. The positive surface charges of Hombikat UV100 TiO2 and Aeroxide P25 TiO2 can create favorable conditions for anionic oxalate complexation. However, the complexation of anionic oxalate on the negatively charged surface of WO3 is not favored, resulting in the low positive effect of oxalate on the degradation of ranitidine. As expected, the adsorption of oxalate on the Hombikat UV100 TiO2 surface was significantly higher than on the WO3 surface (Fig. 6b). This behavior confirms that when the metal oxide possesses a more positive surface charge, the positive effect of oxalate on the degradation process in the metal oxide/H2O2 system is more pronounced by enhancing the complexation of anionic oxalate.
The positive effect of oxalate in the Hombikat UV100 TiO2/H2O2 system was observed under acidic conditions (i.e., krel > 1.0 at pH 3.0 and 5.0); however, the effect of oxalate was negative under basic conditions (i.e., krel < 1.0 at pH 9.0 and 11.0) (Fig. 6c). This phenomenon is also associated with the pH-dependent surface charge of TiO2. When the suspension pH is lower than the PZZP of Hombikat UV100 TiO2 (pH 6.0), the surface charge becomes more positive, thereby enhancing the complexation of oxalate as the pH decreases. Therefore, the extent of the positive effect of oxalate on ranitidine degradation at pH 3.0 (krel = 7.679) was greater than that at pH 5.0 (krel = 5.715). However, the surface charge becomes negative when the suspension pH is higher than the PZZP of Hombikat UV100 TiO2. At pH 9.0 and 11.0, the electrostatic repulsion between anionic oxalate and the negatively charged surface banishes the positive effect of oxalate on ranitidine degradation by preventing its complexation. Under these circumstances, the function of oxalate as a scavenger for HO2• and 1O2 inhibits the degradation of ranitidine, leading to krel values lower than 1.0 (krel = 0.698 at pH 9.0 and krel = 0.452 at pH 11.0).
4. Conclusions
Oxalate is a common compound found in wastewater originating from both domestic and industrial sources [46, 47]. Therefore, the effect of oxalate is inevitably observed in wastewater treatment processes. In addition, oxalate can be easily degraded in conventional biological processes [48, 49], making the addition of oxalate to the TiO2/H2O2 system feasible for the degradation of poorly biodegradable ranitidine. In this study, we investigated the effect of oxalate on the degradation of ranitidine in the TiO2/H2O2 system. The presence of oxalate significantly enhanced the degradation of ranitidine by increasing the generation of HO2•. In the TiO2/H2O2/oxalate system, HO2• is generated through two pathways: inner-sphere electron transfer from the –OOH groups to the metal centers in the peroxo metal complexes (i.e., >TiIV–OOH) and the subsequent reduction of oxygen by the complexes formed between oxalate and >TiIII (i.e., >TiIII(oxalate)n(2n–3)−). Among various chelating agents, oxalate showed the highest enhancement in the degradation of ranitidine. In addition, the strategy of adding oxalate can be applied not only to the TiO2/H2O2 system but also to other metal oxide/H2O2 systems to enhance the degradation efficiency of ranitidine. Considering the low material and chemical costs, absence of energy requirements, and high degradation efficiency, the metal oxide/H2O2/oxalate system, particularly TiO2, can be proposed as a viable option for the degradation of ranitidine.