1. Introduction
Electrochemical desalination technology is attracting much attention due to its mild operating environment compared to conventional desalination processes that require high pressure levels or high temperatures [1,2]. Capacitive deionization (CDI) refers to an electrochemical desalination technology in which electrical energy is applied to an electrode to remove charged ions by adsorbing them on the electrode surface. CDI, which can reuse the stored electrochemical energy during its operation, has significant energy advantages over other electrochemical desalination processes. While CDI shows performance comparable to that of RO, a type of commercial desalination technology, in brackish water of 50 mM or less, it is difficult to use in highly concentrated salt water due to the capacity limit of the electrodes [3–5]. To overcome this limit, various new CDI systems, including membrane capacitive deionization (MCDI) [6,7], hybrid capacitive deionization (HCDI) [8–12], and battery deionization (BDI) [13–15], have been proposed. Despite these efforts, the CDI system described above is inefficient because it does not allow ion removal during the discharging step, when ions are released from the electrode.
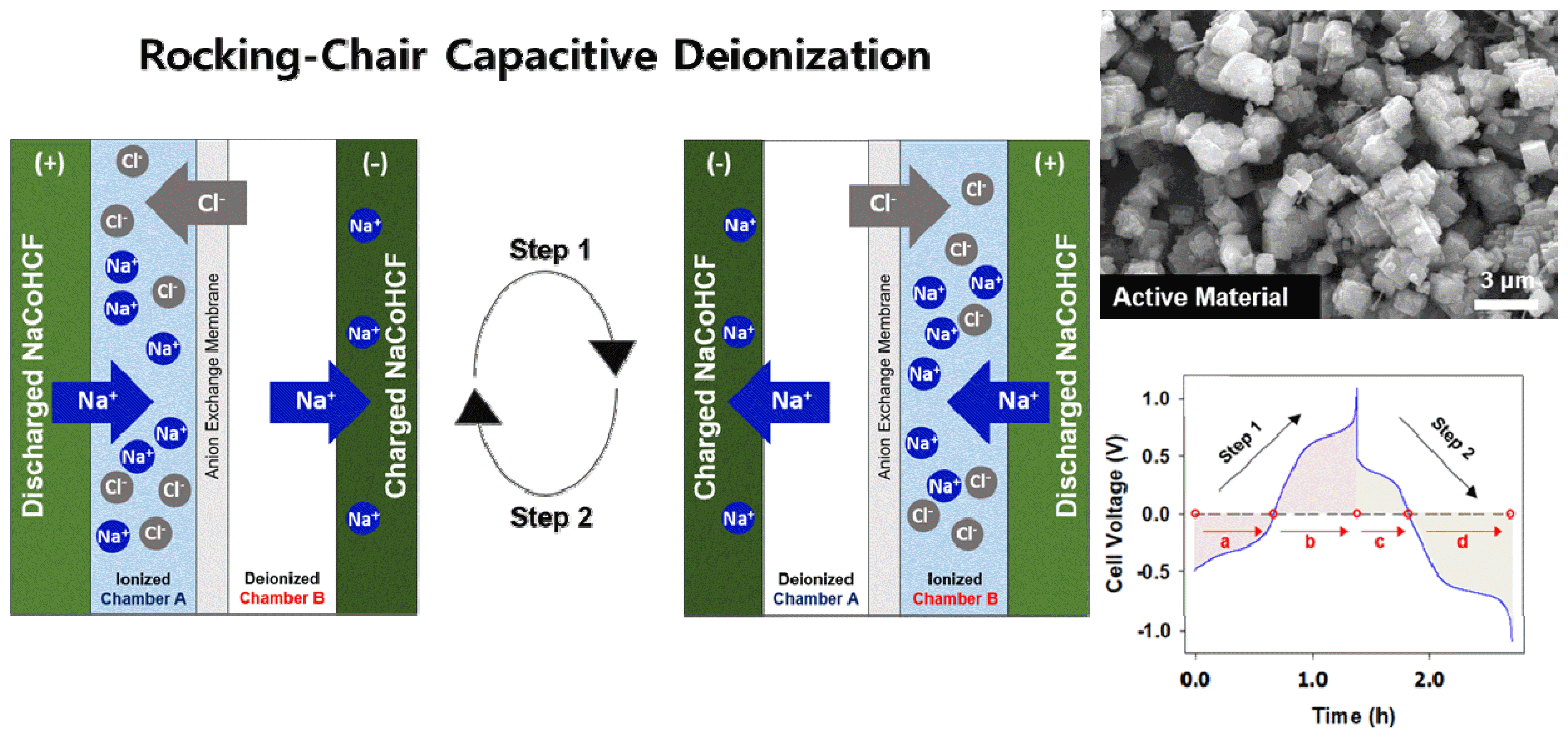
The invention of a cation intercalation desalination (CID), referred as a rocking-chair capacitive deionization (RCDI) by the category of CDI cell architecture [16], significantly increased the system efficiency of CDI by allowing ions to be removed even during the discharging step [17]. In the RCDI system, shown in Fig. 1, two identically sized chambers are formed with two identical cation intercalation electrodes and a membrane positioned at the center of the electrodes. During the operation of the system, the charged electrode captures cations (desalination) while the discharged electrode in another chamber releases cations (concentration). Anions pass through an anion exchange membrane by diffusion in order to compensate for the charge imbalance caused by the movement of cations. Because RCDI can carry out desalination and concentration simultaneously in a single stage, desalination takes place during both stages, i.e., charging and discharging, which increases the efficiency of the entire system [18]. These advantages of the system have led to research on applications of various aqueous faradaic electrodes to RCDI [19–22].
Here, we propose a method to enhance the desalination capacity of RCDI by using sodium cobalt hexacyanoferrate (NaCoHCF) electrodes that utilize two redox active sites. Through an electrochemical analysis, it was determined that the two redox sites of the synthesized NaCoHCF electrode worked properly. In this study, two NaCoHCF electrodes were applied to RCDI and electrochemical desalination was successfully accomplished with the two redox active sites of the NaCoHCF electrodes at a cell voltage range between 0.3V and 1.1V. Furthermore, in order to measure the change in the desalination performance of RCDI using two redox active sites, changes in the desalination capabilities were compared by operating the system with NaNiHCF electrodes using one active site under an identical condition.
2. Materials and Methods
2.1. Electrode Preparation
The active materials of the NaCoHCF electrodes were synthesized via a citrate-assisted controlled crystallization method [23]. Here, 100 mL of a 0.05 M CoCl2 and 0.35 M sodium citrate solution was mixed with 100 mL of a 0.05 M Na4Fe(CN)6 solution under vigorous stirring for 24 hours. To synthesize the NaNiHCF particle, 100 mL of a 0.05 M NiCl2 was added into 100 mL of 0.35 M Na-citrate and 0.05 M Na4Fe(CN)6 mixed solution under vigorous stirred condition. The obtained solutions were aged for 24 hours, and the precipitates were collected by filtration. During the filtration step, the precipitate was washed twice with 200 mL of distilled water and once with 200 mL of ethanol. The obtained active materials were dried in a 60°C oven overnight.
To fabricate the electrodes used in this study, 80 wt% of the prepared active materials, 10 wt% of a conductive material (Super P, Timcal, Switzerland), and 10 wt% of polytetrafluoroethylene (PTFE, Sigma-Aldrich, USA) were gently mixed with ethanol for 20 minutes. The prepared mixture was placed on a roll presser to create a sheet-type electrode approximately 250 μm thick. The prepared sheet-type electrode was dried in a vacuum oven at 120°C overnight to remove the residual solvent.
2.2. Characterization of the Electrode
The morphologies and elemental distribution of the prepared NaCoHCF were characterized by a field emission scanning electron microscope (FESEM, JEOL JSM-7900F, Japan). X-ray diffraction (XRD, Rigaku, Japan) was used to analyze the crystal structure of the prepared NaCoHCF in the 2θ range of 10° – 70° with a ramping step of 1° min−1.
The electrochemical properties of the NaCoHCF electrode were assessed in a three-electrode configuration. In this cell, a 1.0 cm × 1.0 cm NaCoHCF electrode attached to a titanium current collector served as the working electrode, an electrochemically prepared 2.0 cm × 5.0 cm AgCl electrode was used as the counter electrode, and a commercial Ag/AgCl electrode (in Saturated KCl) was used as the reference electrode, with a 1.0 M NaCl solution used as the electrolyte. Cyclic voltammetry (CV) scans were obtained using a potentiostat (PARSTAT 2273, Princeton Applied Research, USA) operated at a scan rate of 0.1 mV s−1. A galvanostatic charge/discharge test was conducted from 0.05 V to 1.10 V under a constant current condition (± 0.1 A g−1) using a battery cycler (WBCS3000, WonATech, Republic of Korea).
2.3. Cell Construction
The two 2.0 cm × 2.0 cm area of NaCoHCF or NaNiHCF electrodes were attached onto each titanium current collector (thickness: 200 μm, Sigma-Aldrich, USA) with carbon paint (DAG-T 502, Ted Pella, USA). To ensure that one of the prepared electrodes was in a fully charged while the other was in a discharged state, one was discharged to −0.2 V (Na+ intercalated) and the other was charged to 1.1 V (Na+ de-intercalated) for 30 min in a 1.0 M NaCl solution using a three-electrode configuration with an Ag/AgCl (in saturated KCl) reference electrode.
A rocking-chair capacitive deionization (RCDI) cell was employed in this experiment. This cell consists of the prepared fully discharged and charged electrodes, two polyamide woven spacers, and an anion exchange membrane (AMV, Selemion, Japan). It is divided into two chambers, denoted here as chamber A with the fully discharged electrode and chamber B with the fully charged electrode, according to the positioning of the anion exchange membrane. The cell was covered by two PTFE and silicone gaskets to prevent the solution from leaking.
2.4. Desalination Tests
A volume of 0.4 mL of a 500 mM NaCl solution was placed in each chamber (total volume of solution in the cell: 0.8 mL). This cell was operated under constant current operation (± 1.0 mA cm−2) over a voltage range of −1.1 V to 1.1 V using a battery cycler (WBCS3000, WonATech, Republic of Korea). After the cell voltage reached the limit voltage, the solutions in both chambers were refreshed with new NaCl solution in amounts of 500 mM. The cation concentration of the extracted solution after each step was measured by means of ion chromatography (ICS-1100, Thermo Fisher Scientific Inc., USA).
The electrochemical desalination performance parameters were calculated based on the results of a full cycle, defined as charging and discharging steps. The ion removal efficiency was calculated as follows:
The salt adsorption capacity (SAC) was calculated using the Eq. (2).
where M.WNaClis the molecular weight of NaCl (58.4 g mole−1), me is the total weight of both electrodes including the Super P and PTFE (g), ΔCi is the concentration change of step i (mM), and Vc is the volume of the chamber used (mL).
The charge efficiency (Λ) was calculated as Eq. (3).
where F is the Faraday constant, zi is the valance state of i, Δni is the molar change of i, and ∑ is the transferred total charge.
The specific energy consumption (SEC) was calculated via Eq. (4).
where I is the applied current, m is the mole of the removed NaCl, tcycle is the total operation time, and Vcell is the cell voltage during operation.
3. Results and Discussion
3.1. Electrode Characterization
Fig. 2 shows the crystalline structure and morphology of the prepared NaCoHCF electrode.
As shown in Fig. 2(a), the XRD diffraction line of the synthesized NaCoHCF is clear and in good agreement with those in previous studies, indicating that NaCoHCF with highly crystalline face-centered cubic (FCC) structure was synthesized. Moreover, the uniformly distributed C, N, O, Na, Fe and Co elements in the energy dispersive spectroscopy (EDS) mapping images support the synthesis of high-purity NaCoHCF (Fig. 2(b)). In a detailed comparison with the standard diffraction pattern of Prussian blue (JCPDS 52-1907), it can be seen that the lines are split by doublets at 220, 420, 440, and 620, which means that PBA is in a rhombohedral state. The result of the rhombohedral state reveals that the synthesized NaCoHCF material is in a state where two sodium ions are intercalated within the PBA structure. Typical PBA has a cubic structure, but it is known that when secondary sodium is inserted into the PBA frame, the cubic structure is distorted and changes to a rhombohedral state [23].
As can be seen in the FESEM image in Fig. 2(b), a highly crystalline NaCoHCF material of approx. 5 μm was synthesized. These synthesized particles were larger than regular PBAs, with particles as small as 50 nm. While the PBA particles prepared by the conventional method are small in size due to their fast reaction rate and low solubility, we used a kinetically controlled crystallization method to synthesize NaCoHCF with a higher order and a larger particle size. This method was reported in a previous study using Na-citrate as a chelating agent to slow the reaction rate through the slow release of Co2+. Finally, a sheet-type electrode with a thickness of approx. 200 μm was well fabricated. Through a FESEM analysis, for the fabricated sheet-type electrode, it was additionally confirmed that the active material (NaCoHCF), the conductive material (Super P), and the binder (PTFE) were well mixed, as shown in Fig. 2(c).
3.2. Electrochemical Characterization
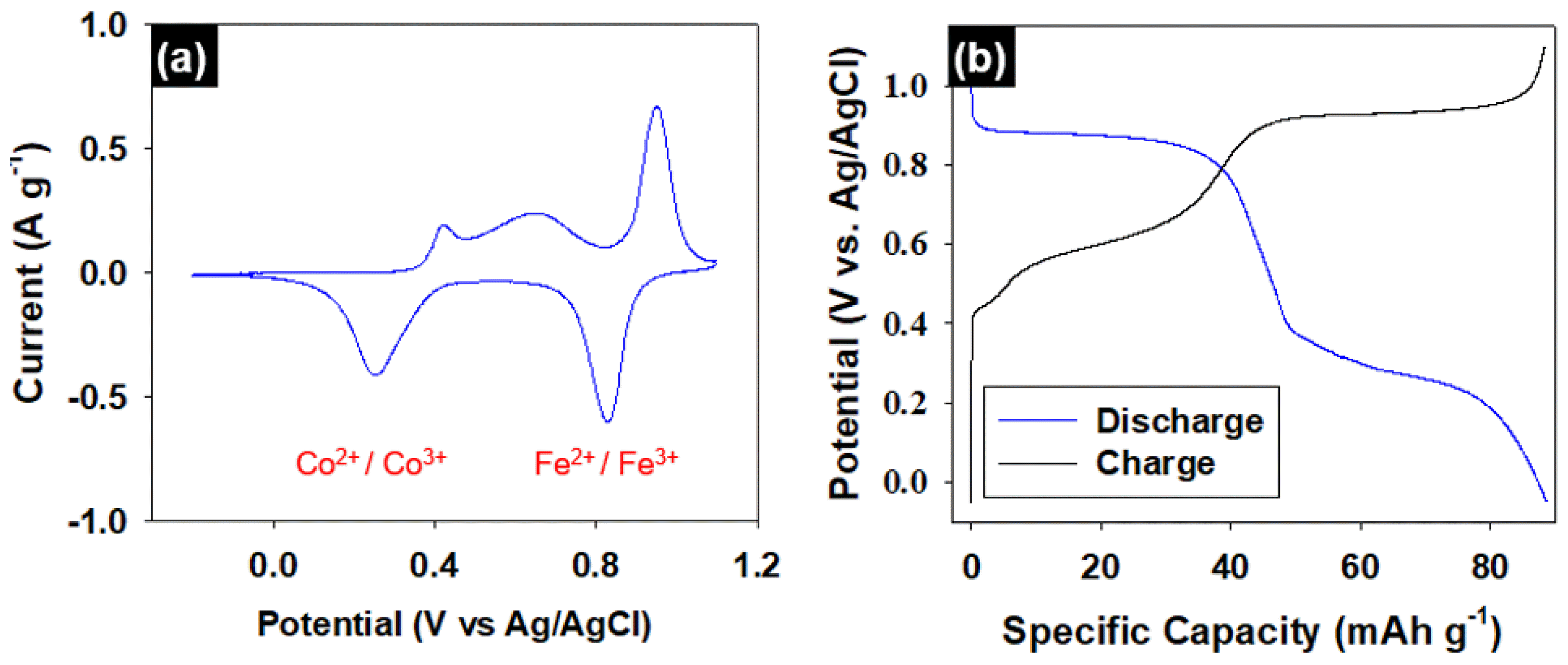
To clarify the electrochemical properties of the NaCoHCF electrode further, cyclic voltammetry (CV) and galvanostatic charge/discharge testing were conducted with a 1.0 M NaCl solution and a three-electrode configuration (Fig. 3). As shown in Fig. 3(a), the CV curve presents two distinguishable oxidation/reduction peaks at 0.4 V and 0.9 V. The shape of this curve is the typical CV curve of intercalation materials, which demonstrates that Na+ can be reversibly de-/intercalated. More importantly, the presence of two redox peaks indicates that the electrode follows the 2-Na reaction mechanism. The 2-Na reaction mechanism of NaCoHCF originates from two-redox active site reactions of the Co2+/Co3+ and Fe2+/Fe3+ couples at 0.4 V and 0.9 V, respectively. The charge-spin lattice strength capabilities of the two transition metals differ due to the fact that Co is nitrogen-coordinated and Fe is carbon-coordinated, which causes the two redox active sites to have different operating potentials [24]. This redox potential differs significantly from the typical electrode potential of Fe(CN)64−/Fe(CN)63−, and is even higher than that of the Co2+/Co3+ couple, due to the unstable condition of the Fe2+/Fe3+ couple in the Co–Fe framework [25]. In addition, the Na+ insertion/extraction reaction during the Co2+/Co3+ couple can lead to a significant volume fluctuation to accommodate the structural change and this broad peak between 0.4 to 0.65 V expected to show by the volume changes [26]. Consequently, in the Co-Fe framework Prussian blue, the redox potential of Fe2+/Fe3+ is higher than that of Co2+/Co3+ pair.
Fig. 3(b) shows the galvanostatic charge/discharge profile at a current density of 0.1 A g−1 in the 1.0 M NaCl solution. It was confirmed that the potential profile had two plateaus at 0.4 V and 0.9 V. This result is consistent with the CV results, implying that the NaCoHCF electrode follows the 2-Na reaction mechanism at a current density of 0.1 A g−1. This 2-Na reaction mechanism leads to the very high specific capacity of 88 mAh g−1 in this case (active material: 110 mAh g−1). This specific capacity of the NaCoHCF electrode is the highest among faradaic electrodes applied to electrochemical desalination thus far, as shown in Table 1. In addition, the Coulombic efficiency, representing the ratio of the discharge capacity and the charge capacity, is 99.7%, indicating that the NaCoHCF electrode operates reversibly without accompanying side reactions such as water decomposition. Consequently, the properties of the NaCoHCF electrode with superior specific capacity and reversibility suggest that it is a promising cation-reactive electrode material for CDI.
3.3. Desalination Performance
Fig. 4 shows the desalination performance of RCDI using the NaCoHCF electrodes under constant current operation at a current density of 1.0 mA cm−2 in a 500 mM NaCl solution. As shown in Fig. 4(a), the system operates reversibly in the cell voltage range of −1.1 V to 1.1 V in the 500 mM NaCl solution. During the charging and discharging steps, the cell voltage profile of this system gradually changed in the range of −0.9 V to 0.9 V, whereas it changed rapidly at both ends. This result implies that the full capacity of the NaCoHCF electrodes is utilized at the set cell voltage limit. The potential profile of each electrode demonstrates that the electrode uses its full capacity at the set cell voltage limit. The measured potential profiles of the both electrodes have two plateau regions at 0.4 V and 0.9 V, meaning that the aim of this study was achieved by using the two redox active sites of the NaCoHCF electrodes within the set cell voltage range.
Fig. 4(b) shows the resulting ion concentration changes after each step. As described above, an important characteristic of RCDI is that desalination and concentration occur simultaneously in one step. In step 1, concentration occurs in chamber A and desalination occurs in chamber B. Step 2 works exactly the opposite of step 1. After the completion of steps 1 and 2, the Na+ concentrations in chambers B and A were 55 mM and 68 mM, resulting in 88% salt removal on average in 500 mM of saline during one cycle. This demonstrates close to a 10% increase in salt removal compared to the rates in previous studies that utilized Ag/AgCl electrodes, which to the best of our knowledge represents the highest salt removal rate of any CDI system applied to saline concentrations above 500 mM [15]. The high salt removal rate of 88% is attributed to the high specific capacity of the NaCoHCF electrodes (88 mAh g−1) stemming from the use of two redox active sites.
3.4. Desalination Performance Comparison
Fig. 5 shows a comparison of the desalination performance outcomes of the NaCoHCF cell and the NaNiHCF cell at 1 mA cm−2 in 500 mM of NaCl. To ensure a fair comparison of the system performance changes due to the introduction of the NaCoHCF electrode, NaNiHCF, one of PBAs applied to a typical CDI system, was operated under identical conditions apart from the cell voltage range. The cell voltage ranges were set differently because the working potential ranges of the NaCoHCF (0.4 – 0.9 V vs. Ag/AgCl) and NaNiHCF (0.5 – 0.7 V vs. Ag/AgCl) electrodes are different. Fig. 5(a) confirms that the cell capacity per unit electrode weight of the NaCoHCF cell (272 C g−1) was approximately 1.5 times higher than that of the NaNiHCF cell (180 C g−1). The higher specific capacity of the NaCoHCF electrode is attributed to the use of two redox potential regions of the NaCoHCF electrode. The four plateau regions of the NaCoHCF cell in the cell voltage profile in Fig. 5(a) indicate that two redox potential active sites were used in both charge/discharge steps. This result is well supported by the voltage potential profile of each electrode in the NaCoHCF cell. On the other hand, the voltage profile of a NaNiHCF cell with two plateau regions in the charge/discharge steps implies that one redox active site was employed.
Fig. 5(b) compares the changes in the quantitative electrochemical desalination performance capabilities of the cells with the NaCoHCF electrodes and the NaNiHCF electrodes. The NaCoHCF cell achieved approx. 88% ion removal and a SAC value of 123 mg g−1, representing a 48% improvement in the ion removal efficiency and a 43.7% higher SAC value compared to the NaNiHCF cell. This result indicates that the NaCoHCF cell not only allows a longer operating time compared to the NaNiHCF cell but that the longer operating time was in fact used to increase the desalination capacity. In addition, it was confirmed that the charging efficiency, which is the ratio of the amount of charge actually used for desalination to the amount of charge applied to the system, was higher in the NaCoHCF cell (88.8%) than in the NaNiHCF cell (82.5%). This result means that the NaCoHCF electrode applied to the RCDI cell is a very efficient for desalting highly concentrated saline water. In particular, the SAC value of 123 mg g−1 (active material: 154 mg g−1) of the NaCoHCF cells is the remarkable value in relation to CDI, as shown in Table 1. The high SAC value of the NaCoHCF system is due to the high capacity of the NaCoHCF electrode and the continuity at which desalination can occur in both steps 1 and 2, a unique characteristic of the RCDI system [20].
Although the NaCoHCF cell showed the most improved desalination performance compared to the NaNiHCF cell, the specific energy consumption increased by 2.5 times. The NaCoHCF cell has a higher SEC value than the NaNiHCF cell because the resistance of the electrolyte increases as the ion removal rate increases. The final concentration of the NaCoHCF cell is approx. 60 mM, whereas the final concentration of the NaNiHCF cell is approx. 200 mM. Accordingly, it can be seen that the solution resistance of the NaCoHCF cell is much larger. This result is also well supported by the voltage drop difference results between the NaCoHCF cell and NaNiHCF cell. A voltage drop occurs when going from step 1 to step 2 in the cell voltage profile. The voltage drop of NaCoHCF cell was 0.63 V, which is 2.5 times higher than the voltage drop of the NaNiHCF cell (0.25 V). The main causes of the cell voltage drop are known to be polarization through the solid-liquid interface, the resistance of the electrode and ion exchange membrane, and the electrolyte resistance. In this case, the difference in the voltage drop between the two cells is expected mainly to stem from the electrolyte resistance according to the difference in the ion removal rate. It is a fact that the operation condition of current density (1.0 mA cm−2) is not enough for sufficient rate performance compared with conventional CDI systems, and that is one of the main limitations of the RCDI using battery electrode materials [18]. Given that an increase in SEC according to an increase in the ion removal rate is inevitable, it is considered that a follow-up study to find the optimal condition between the ion removal rate and SEC value is required.
4. Conclusions
This present study demonstrates that NaCoHCF electrodes with two redox active sites can be applied to electrochemical desalination to increase the desalination capacity in an RCDI system. The system exhibited 88% ion removal in a 500 mM NaCl solution and achieved a desalination capacity of 123 mg g−1 (active material: 154 mg g−1). Compared to the NaNiHCF electrode using one redox active site under identical conditions, the ion removal rate and desalination capacity were improved by 48% and 44%, respectively. These achievements come from the specific capacity of the NaCoHCF electrode given its use of two redox active sites, which is 1.5 times higher than the specific capacity of a NaNiHCF electrode that uses only one redox active site. Overall, this study proposes that the NaCoHCF electrode with two redox active sites could be a good CDI electrode candidate due to its high desalination capacity.