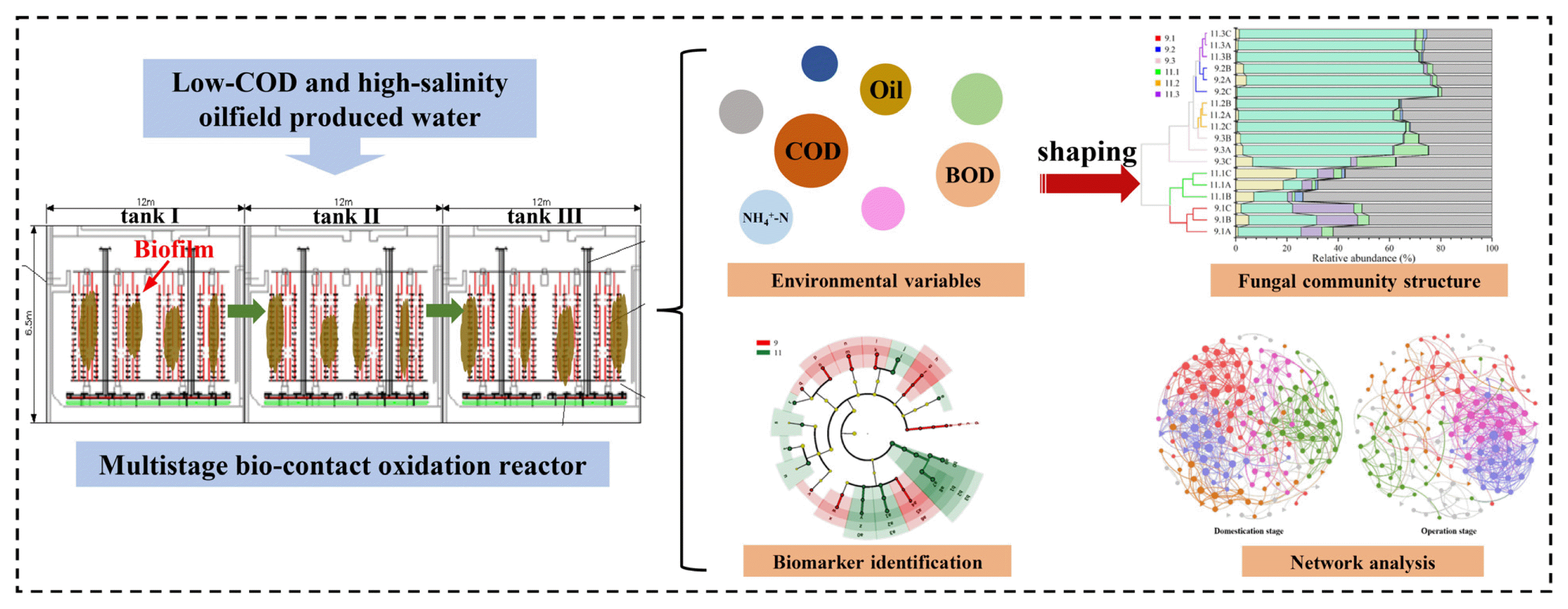
1. Introduction
The large volume of oilfield wastewater is one of the major environmental concerns due to its inclusion of various toxic compounds such as heavy metals, alkanes, BTEX (i.e., Benzene, Toluene, Ethylbenzene, and Xylene), polycyclic aromatic hydrocarbons (PAHs), ammonia nitrogen, and sulfide, posing potential hazards to human health [1]. Conventional physical and chemical methods used to address this problem could be cost-intensive and energy-consuming [2]. The use of microorganisms for bioremediation and detoxification of wastewater is considered as an efficient and lucrative alternative [3]. By using microbial systems, ecologically safe and environmentally friendly technologies can be designed to alleviate pollution.
Among the biological strategies for wastewater treatment, bio-contact oxidation (BCO) is a commonly used technology and is characterized as energy-efficient and cost-effective [4]. Of this system, contaminants were removed and biodegraded through the thorough contact of the biofilm, which was attached on the carriers and formed when the aerated wastewater flowed through the carriers [5]. Successful applications of the BCO reactor in oily wastewater treatment have been shown in previous literatures. For instance, Lu et al. [6] reported that the application of a hydrolysis acidification/BCO system effectively reduced the chemical oxygen demand (COD), ammonia nitrogen, total suspended solid, and total petroleum hydrocarbons in high-salinity oilfield-produced water. Similarly, a study conducted by He et al. [7] showed that the aerobic BCO reactor efficiently removed most of the investigated pollutants from oily wastewater. These studies proved BCO as a promising approach in the bio-treatment of oilfield wastewater.
To date, most studies related to wastewater bioremediation high-lighted the role of bacteria in pollutants degradation. Nevertheless, fungi are also potentially important contributors in various processes during the remediation and have been reported to contain a series of enzyme systems involved in the degradation, especially the well-known cytochrome P450 (CYP450) enzyme systems [8, 9]. They can participate in multiple pathways in the wastewater treatment systems, such as organic compounds degradation [10] and sludge flocs construction [9]. Some white rot fungi can produce extracellular polymeric substances, which could play a role in the biofilm formation [11]. Besides, fungi generally exhibited stronger tolerance against harsh environment than bacteria and could convert toxic compounds into low toxic and nontoxic end products, providing suitable conditions for bacterial growth [12]. Moreover, the filament of fungi could serve as a carrier for the immobilization of bacteria [13]. Overall, these observations suggest the necessities to comprehensively characterize the role of fungal communities in the wastewater treatment systems.
Currently, the pieces of information on fungal communities in wastewater bio-treatment and pollutants degradation remain vastly unexplored and the functions of fungi are still underestimated. Besides, recent studies have shown the strong ecological linkage and co-occurrence patterns between fungi and bacteria in different environments such as PAH-contaminated soil [14], chicken manure compost [15], and tillage [16]. A deep understanding of their interactions will provide critical information and instructive guidance for the modification and improvement of the performance of pollutants removal to meet the increasingly strict wastewater discharge standard. Nevertheless, few studies have been conducted to investigate the interconnection of fungal-bacterial communities in a multistage BCO reactor (MBCOR) during the treatment of oily wastewater, especially for the wastewater characterized by both low COD and high salinity, which could greatly inhibit the degradation process.
Since biofilms play major roles in decomposing pollutants in MBCOR, the analysis of microbial communities within biofilm samples is essential to uncover the bioremediation mechanisms. The rapid development of sequencing technologies provides convenience for the exploration of microbial community in wastewater treatment systems [17]. Our previous study has revealed the significant role of bacterial community within a MBCOR, which was located in Karamay, Xinjiang Uyghur Autonomous Region (45.68° N, 85.05° E), in treating low-COD and high-salinity oilfield wastewater [18]. In this study, we investigated the shift of fungal community in biofilm samples during the wastewater treatment through Illumina high-throughput sequencing. Meanwhile, the feature fungal taxa at different stages were analyzed via linear discriminant analysis (LDA) effect size (LEfSe) analysis. Furthermore, the interactions of fungal and bacterial communities were explored through network analysis. To the best of our knowledge, this study is the first to evaluate the fungal community succession in a MBCOR during the treatment of low-COD and high-salinity oilfield wastewater, providing useful information for deepening the understanding of pollutants removal in such wastewater treatment systems.
2. Materials and Methods
2.1. Descriptions of MBCOR and Analytical Methods
The investigated MBCOR began to work on September 7th, 2018. The first two months were domestication stage which allowed the microorganisms to adapt to the environment of oilfield wastewater. Some basic characteristics of the wastewater were shown in Table S1. It was clearly observable that the wastewater possessed the properties of low-COD (249.3±45.6 mg/L), high-salinity (12969.3±1058.6 mg/L), and high-temperature (47.9±1.6°C). The schematic diagram of the MBCOR was shown in Fig. S1. Detailed operational processes of the MBCOR have been described in our previous study [18]. After acclimation, the MBCOR was continuously operated for 181 days under aerobic conditions at ambient temperature. Wastewater samples were collected throughout the operation process, transferred to sterile plastic tubes immediately after sampling, and preserved in a refrigerator (4 °C). COD, biochemical oxygen demand (BOD), oil content, and ammonia nitrogen in influent and effluent were analyzed by chlorine emendation method, dilution method, infrared spectrophotometry, and Nessler’s reagent colorimetry, respectively, as described previously [18].
2.2. Biofilm Sampling and DNA Extraction
A total of 18 biofilm samples were obtained, which were collected from different tanks (i.e., tank I, II, and III), depths (i.e., 1.5 m, 3.0 m, and 4.5 m), and stages (i.e., domestication stage and operation stage). The sampling of biofilm for domestication stage and operation stage was performed on day 10 and day 75, respectively. The collected biofilms could representatively manifest the feature of fungal community at these stages, as reflected by the discrepancy of pollutants removal performance. The biofilm samples were transferred to sterile glass bottles immediately after sampling and stored in a −80 °C freezer before further analysis. Total fungal genomic DNA was extracted using E.Z.N.A.® Water DNA Kit (Omega Bio-Tek, Norcross, USA) following manufacturer’s instructions. The DNA quality was checked by the ratio of absorbance at 260 nm and 280 nm using a NanoDrop 2000 spectrophotometer (NanoDrop Inc., Wilmington, DE) and 0.8% agarose gel electrophoresis.
2.3. Illumina High-throughput Sequencing
The extracted DNA was then used as the template to amplify fungal ITS gene with the primer sets of ITS1F (GGAAGTAAAA GTCGTAACAAGG) and ITS1R (GCTGCGTTCTTCATCGATGC). PCR products were purified using Cycle Pure Kit PCR (Omega Bio-Tek, Norcross, USA) and paired-end sequenced on an Illumina Miseq sequencing platform (Personalbio Co., Ltd., Shanghai, China) according to the standard protocol. Low-quality Reads and chimera sequences were removed using Trimmomatic and UCHIME, respectively. According to reported literatures, operational taxonomic units (OTUs) were clustered based on a 97% sequence identity threshold using UCLUST algorithm [19–21]. Alpha diversity indices including Good’s coverage, ACE, Chao1, Shannon, Simpson, and observed species were calculated using Quantitative Insights Into Microbial Ecology (QIIME) software. The UNITE (release 5.0) database was used as the reference database for fungal OTU classification. The obtained raw reads were deposited in the NCBI Sequence Read Archive database (accession number SRP266535).
2.4. Data Analyses
Principal coordinates analysis (PCoA) based on bray-curtis distance was employed to reveal the fungal community similarities among different biofilm samples. Canonical correlation analysis (CCA) was performed by the genescloud tools (https://www.gene-scloud.cn) to evaluate the relationship between environmental variables and fungal community structure. Spearman correlation analysis was conducted by R (version 3.6.1) using “psych” package. The correlation is significant when P value is below 0.05, while it is extremely significant when P value is below 0.01. LEfSe analysis was applied to detect the potential biomarkers in fungal communities using OmicStudio tools (https://www.omicstudio.cn/tool). Phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt2) algorithm was employed to predict ecological functions of fungi on Bioincloud platform (https://www.bioincloud.tech/). The co-occurrence networks were constructed by Gephi software (version 0.9.2). The topological properties of Erdös–Réyni random networks were calculated by R using igraph, openxlsx, and tidyverse packages. The bacterial community data used for network construction were derived from the same MBCOR as the fungal community and have been reported in our previous study [18].
3. Results and Discussion
3.1. Fungal Community Characteristics
3.1.1. Fungal richness and diversity
The monthly average contents of COD, BOD, NH4+-N, and oil in influent and effluent were shown in Fig. S2. The average removal efficiencies for COD, BOD, NH4+-N, and oil during the operation stage were 65.5%, 64.8%, 81.0%, and 70.2%, respectively, suggesting that biofilms in MBCOR could effectively decompose pollutants comprised in the high-salinity and low-COD produced water [18].
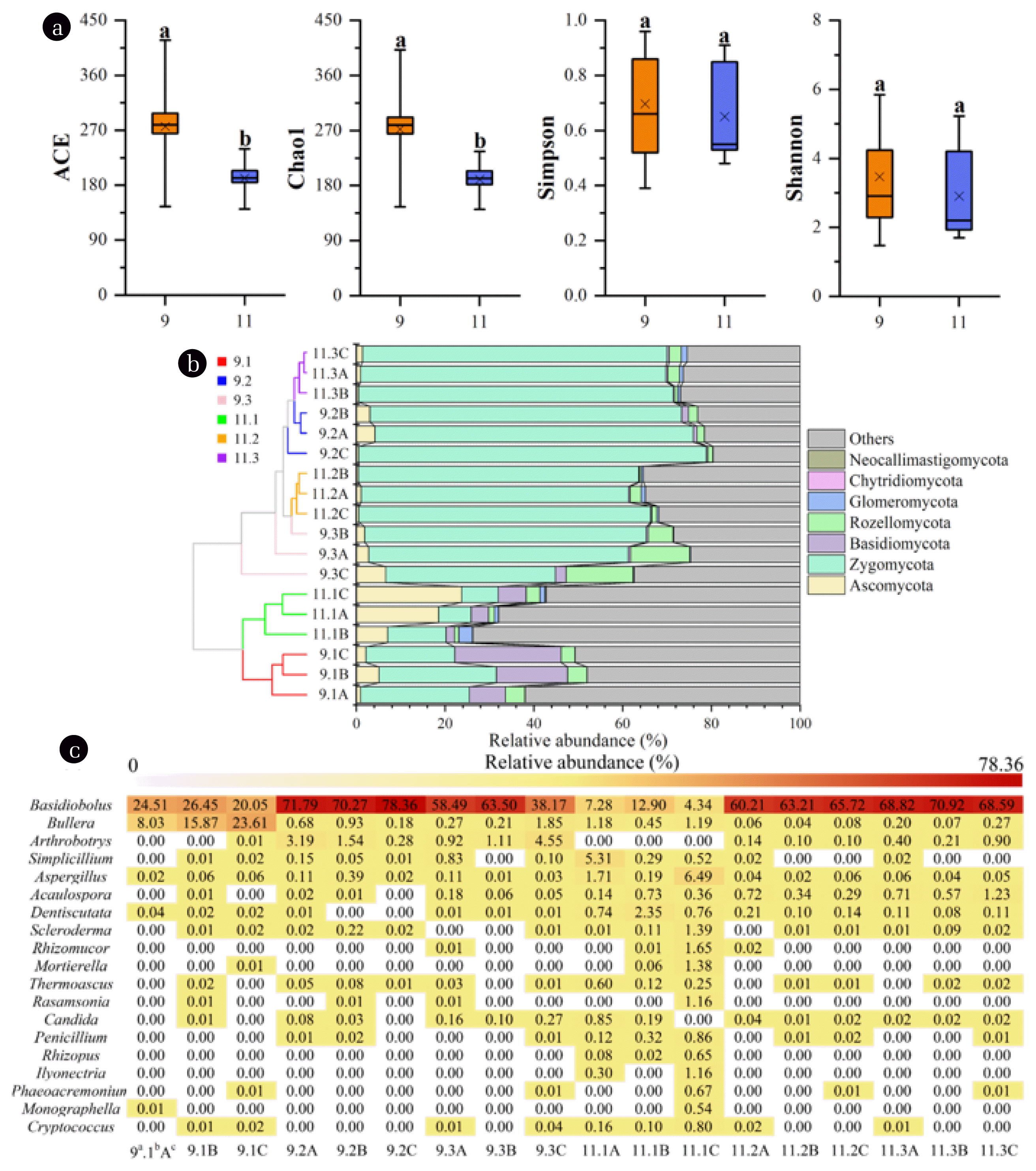
To explore the roles of fungi in pollutants degradation, fungal community structures of different biofilm samples were investigated through high-throughput sequencing. Meanwhile, the reasonability of fungal abundance for wastewater treatment was verified by real-time quantitative PCR. The mean abundance of ITS gene for domestication and operation stage was 2.38±0.51×107 and 7.64±1.62×107 copies/g wet weight biofilm, respectively, which was close to the result reported by Wei et al. [22] obtained from activated sludge. With respect to the result of high-throughput sequencing, a total of 721,454 (ranging from 27655 to 82424) effective sequences for all samples were obtained. The obtained OTUs ranged from 11 to 48 (Table S2). The coverage indices were in range of 0.992–0.998, demonstrating the effectiveness of constructed sequence libraries in covering the fungal communities (Fig. S3). Alpha diversity indices showed that the species richness was significantly (P<0.05) higher at domestication stage than operation stage (Fig. 1a), demonstrating the stabilization of fungal community after acclimatization. Similarly, Shannon and Simpson indices decreased slightly after the domestication stage, indicating the reduction of fungal diversity of the investigated biofilm samples. The result suggested that certain components in produced water played significant roles in shaping the fungal community structure, eliminating maladaptive microorganisms, and simplifying community compositions. A similar pattern was shown in bacterial community from the same MBCOR [18]. Besides, fungal diversity in tank I was significantly higher than other tanks (Table S2), which is probably due to the high pollutants content in this tank, serving as substrates and supporting the proliferation of diverse fungal genera. Moreover, it was found that the depth did not significantly (P>0.05) affect α-diversity indices of microbial community as tested by one-way ANOVA.
3.1.2. Fungal community structure
The fungal taxonomic distributions of different biofilm samples were investigated to explore the shift of community structure. The multistage structure of MBCOR resulted in a differentiation in fungal community (Fig. 1b), especially the tank I and other tanks, which primarily resulted from the discrepancy of environmental conditions in different tanks [18]. Seven major fungal phyla were detected in all samples, among which Zygomycota (7.37%–78.36%) dominated the fungal community. This phylum was reported to be significantly associated with primary and secondary metabolites such as sugar, amino acids, and organic acids [23]. The sharp increase in relative abundance of this phylum in tank II and tank III is probably related to the high concentration of metabolites in these tanks received from tank I. In addition, the proportion of Ascomycota raised from 0.95%–5.11% at domestication stage to 7.04%–23.76% at operation stage within tank I. This phylum is the largest group in aquatic fungal kingdom, contributing for more than 65% of the currently known fungi and key fungal communities for the decomposition of organic pollutants in nature [24]. Similarly, the abundance of Basidiomycota elevated in in tank I at operation stage (Fig. 1b). Members belonged to phylum are also known to mineralize organic contaminants [24, 25]. The enrichment of these phyla in tank I implies the significant role of this tank in transforming the contaminants, which is in accordance with the result obtained in our previous study [18]. Meanwhile, this phylum possesses the feature of heat tolerant [26], which also enabled its proliferation within the wastewater of high temperature (>45°C) in tank I. Moreover, considerable proportions of fungal populations remained unclassified or uncultured, especially for biofilm samples in tank I, suggesting the fungal community was rather complex.
To obtain a better resolution of fungal community, taxonomic distributions at genus level was further investigated. Nineteen major genera were identified, and the result was presented in Fig. 1c. Since the MBCOR was operated under aerobic conditions, most of these fungal populations were aerobes or facultative aerobes. Among the identified genera, Basidiobolus is predominant in the majority of biofilm communities with the relative abundance ranging from 4.34% to 78.36%. This genus is a typical saprobe and usually exists in soil or plant detritus, playing important roles in the decomposition of organic matters [27]. In this study, the high abundance of this genus implied that it might participate in pollutants transformation. Besides, Bullera (8.03%–23.61%) was found to occupy high proportions in tank I at domestication stage. Bullera was reported to be capable of secreting extracellular polysaccharides which are important substrates for biofilm formation [28]. At the operation stage, when the biofilm was mature and strong enough to resist environmental disturbance, the relative abundance of this genus decreased dramatically. In addition, Aspergillus is a halophilic fungus and was affiliated with PAHs degraders, possessing strong abilities in utilizing PAHs and other xenobiotic compounds in saline conditions [29]. Meanwhile, this genus is capable of producing biosurfactant which can assist the hydrophobic pollutants biodegradation [30]. The increase in relative abundance of this genus in tank I at operation stage demonstrated its potential role in reducing oil content (Fig. 1c). Other fungal genera such as Penicillium, Simplicillium, Candida, Rhizopus, and Scleroderma were all previously reported to be associated with hydrocarbons degradation, biosurfactant production, biofilm formation, and ammonia nitrogen assimilation [31–34].
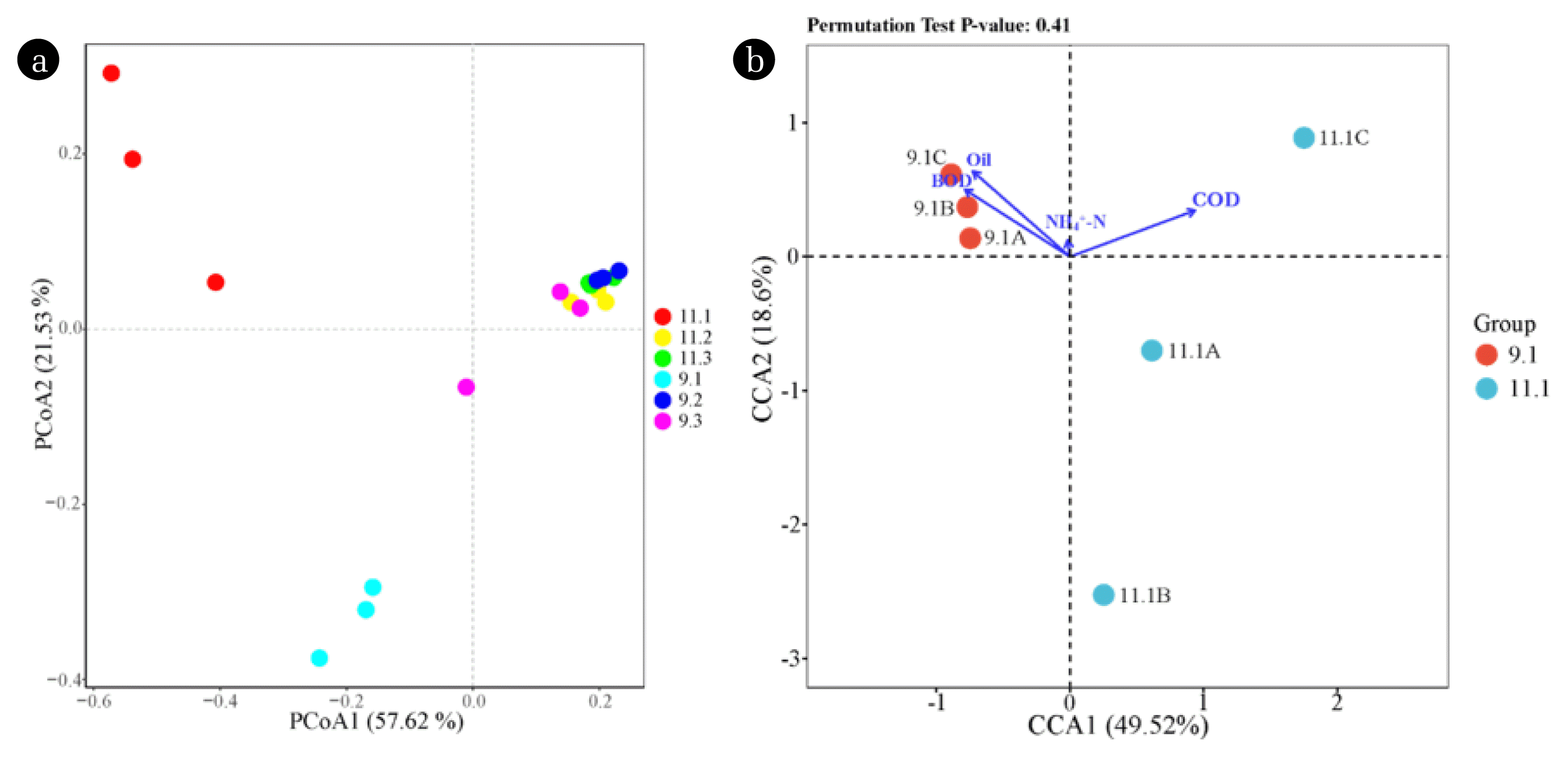
PCoA analysis (OTU level) can reveal the community similarities among different biofilm samples. As shown in Fig. 2a, the axis1 and axis2 of PCoA explained 57.62% and 21.53% of the data distribution, respectively. An apparent fungal community differentiation was observed in biofilm samples between tank I and other tanks. This could be ascribed to the high hydraulic loading and pollutants concentration within the first tank, selectively enriching certain microbial populations that can tolerate harsh environments. An analogous result was observed in bacterial communities [18]. With respect to tank II and tank III, the fungal communities exhibited high similarities, which is probably due to their reception of wastewater with similar constituents. The CCA results further illustrated the importance of environmental factors in shaping the fungal community structure (Fig. 2b). The axis1 and axis2 of CCA explained 49.52% and 18.60% of the microbial community structure, respectively. Apparently, BOD, COD, and oil content were major variables in influencing the fungal communities within tank I, among which COD was the most important factor. A similar result has been reported by Cortés-Lorenzo et al. [35] that COD is one of the most important factor in influencing the fungal community structure within a submerged fixed bed bioreactor during the treatment of urban saline wastewater.
3.2. Biomarker Identification and Functional Profiles Prediction
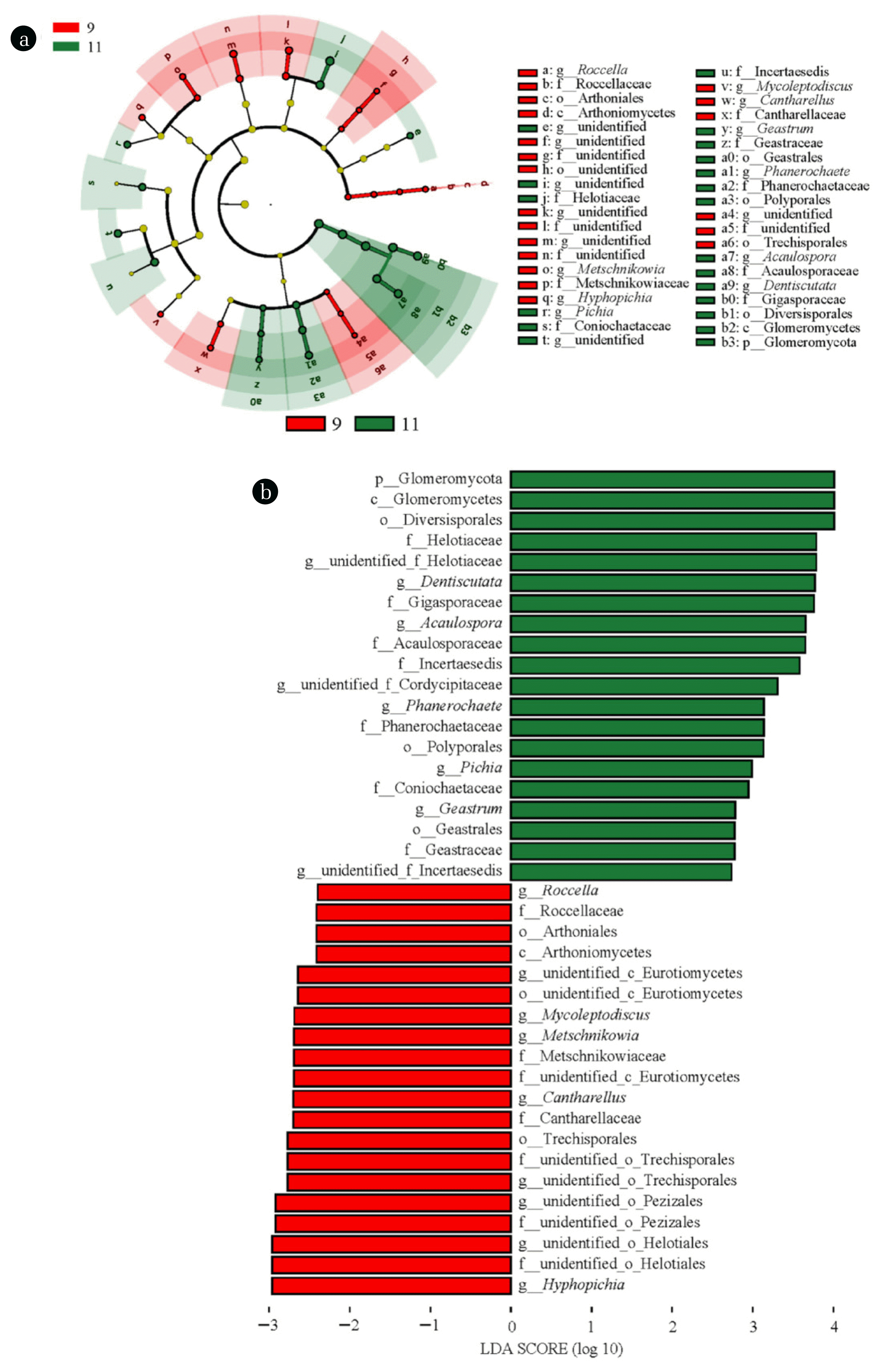
To identify the feature fungal taxa in biofilm samples at different stages, LEfSe analysis was conducted, and the result was presented in Fig. 3. Forty fungal clades presented statistically significant difference (LDA value > 2.0, P<0.05) at two stages. The representative fungal genera with variations in abundance at domestication stage included Cantharellus, Roccella, and Metschnikowia. These genera have been shown to carry genes related to the synthesis of extracellular lipid, protein, and polysaccharides, such as Acc gene encoding acetyl-CoA carboxylase and Fas gene encoding fatty acid synthase, which enables them to play certain roles in biofilm construction and formation [36, 37]. With respect to the operation stage, members including Phanerochaete and Pichia are typical PAH degraders and have been reported to contain CYP450 genes, implying that they might involve in PAHs biodegradation during the wastewater decontamination [38–40]. Besides, Geastrum is capable of producing a variety of exoenzymes such as laccase, manganese peroxidase, and lipase, indicating its potential role in catalyzing the decomposition of organic pollutants [41]. Thus, the feature fungal taxa at different stages suggested the fungal community shift is driven by functional requirements.
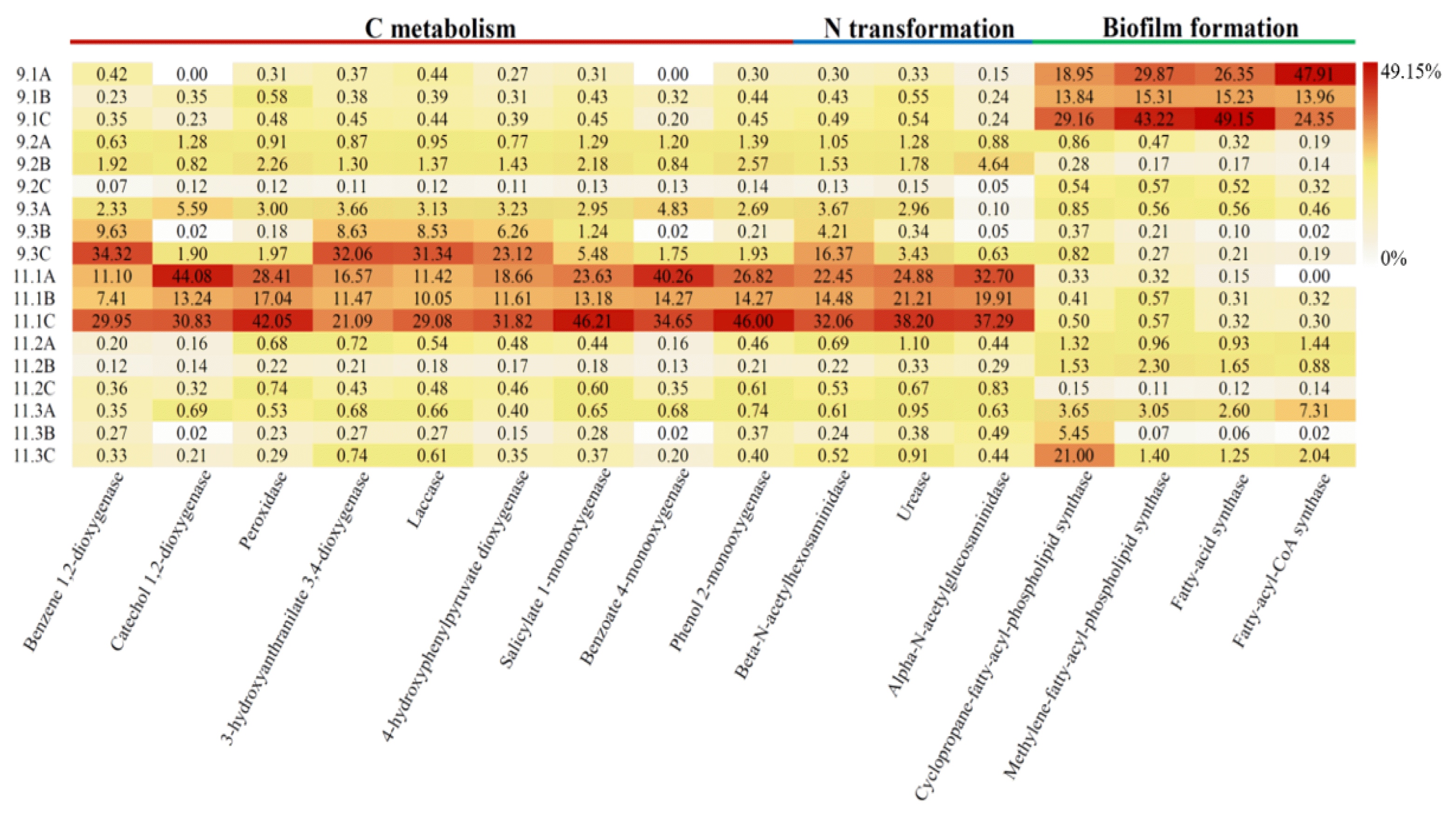
The newly updated tool PICRUSt2 was applied to explore the functional profiles of fungal community in different biofilm samples. Enzymes related to carbon metabolism, nitrogen transformation, and biofilm formation such as peroxidase, laccase, monooxygenase, dioxygenase, and synthase, as reported in previous studies, were selected from the results of functional predictions [42, 43]. An obvious division in fungal function was observed at different stages (Fig. 4). Where, enzymes associated with biofilm construction were greatly enriched at domestication stage, while pollutants degradation related enzymes rose at operation stage. The result was in accordance with the pollutants treatment performance at different stages and confirmed that fungal community succession was function-driven. In addition, the result also implied that microorganisms could adapt to harsh environments by regulating the synthesis the required enzymes.
3.3. Co-occurrence Patterns of Fungal and Bacterial Communities
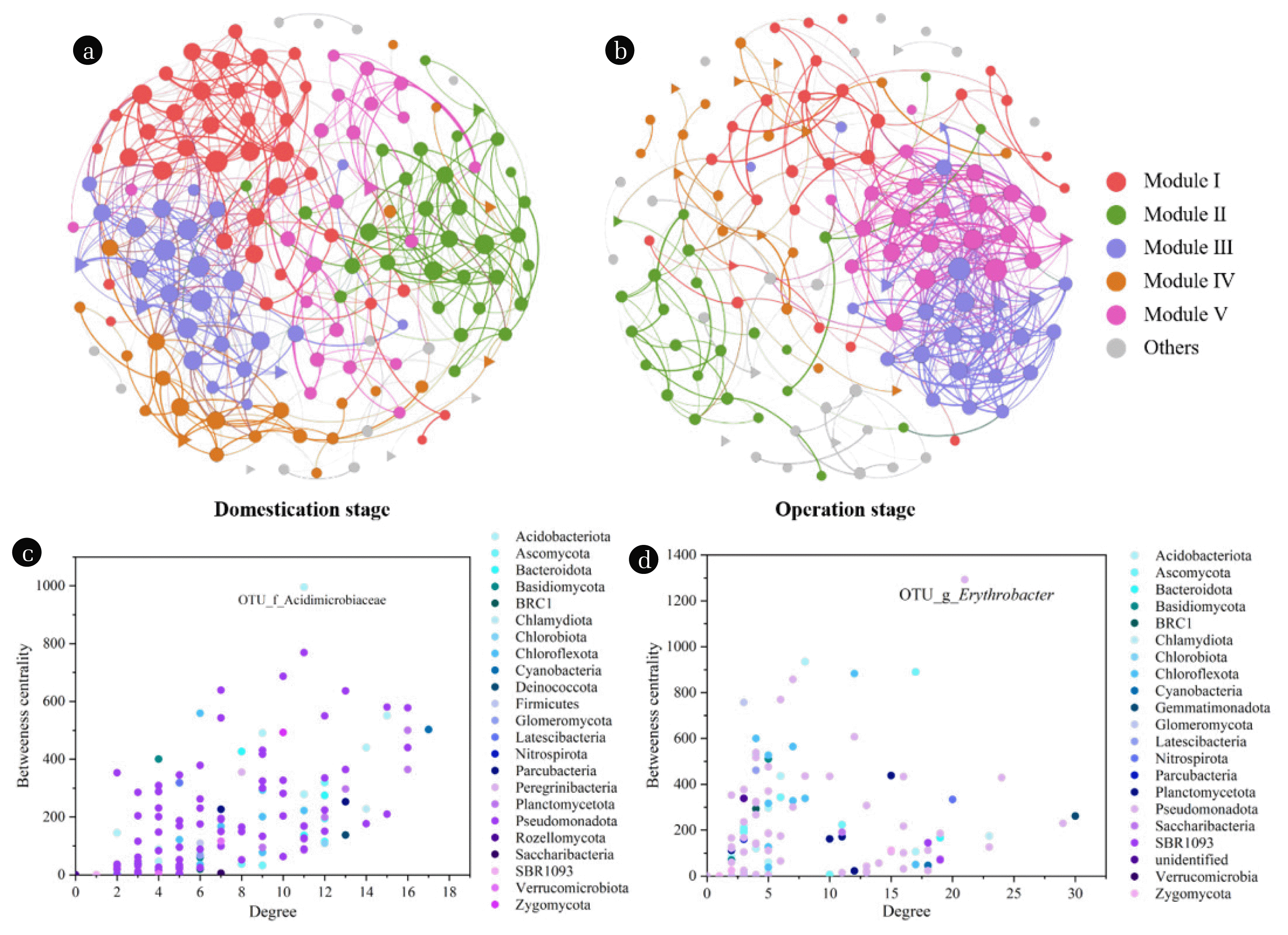
A co-occurrence network was constructed to elucidate the interactions between bacterial and fungal communities at different stages. As shown in Fig. 5, fungal and bacterial genera were highly interconnected and exhibited evident non-random co-occurrence patterns, indicating that fungi and bacteria have a synergistical cooperation during the wastewater treatment. Topological properties of constructed networks were calculated and compared with those of Erdös–Réyni random networks. Properties including clustering coefficient (CC), graph density (GD), modularity (MD), average path length (APL), and network diameter (ND) were all higher than those of the random networks (Table 1). The results suggested that the microbial networks possessed “small world” properties, that is nodes in constructed networks are more connected than in identically sized random networks [44]. Meanwhile, the MD values of networks were both higher than 0.4, suggesting the networks had modular structures [45]. The network at domestication stage (named as “D-network”) consisted of 156 nodes (genera) and 545 edges (367 positive edges), while the network at operation stage (named as “O-network”) comprised 141 nodes (genera) and 496 edges (294 positive edges). In addition, the AD value was slightly higher in O-network than in D-network, indicating a higher network complexity and microbial interactions after biofilm acclimation [46]. A similar pattern was shown in the fungal only networks (Fig. S4). The enhanced interconnections of microbes within and across microbial types could greatly elevate the microbial adaptions and resistance to environmental stress, thus leading to an improved pollutants degradation performance at operation stage [47].
Additionally, both networks contained five major modules, of which modules I–V accounted for 24.4%, 19.9%, 17.3%, 14.7%, and 14.7% of the whole network, respectively, at domestication stage (Fig. 5a). Meanwhile, the five modules were mainly occupied by Pseudomonadota (54.2%), Actinomycetota (7.7%), Chloroflexota (7.0%), and Bacteroidota (6.3%) (Fig. S5a). At operation stage, the microbial linkages were centralized within module III and V, demonstrating that the microbial interactions were enhanced during this process (Fig. 5b). About 18.4%, 17.0%, 17.0%, 14.2%, and 15.6% nodes were distributed in modules I–V, respectively. Pseudomonadota (46.6%), Chloroflexota (10.3%), Actinomycetota (8.6%), Ascomycota (7.8%), and Planctomycetota (6.0%) were the major phyla within these modules (Fig. S5b).
The potential keystone taxa or important nodes are usually identified by betweenness centrality, closeness centrality, degree centrality, and strongly connected ID [48]. In this study, between-ness centrality and degree centrality were used to screen the keystone genera in the fungal-bacterial networks at different stages. As shown in Figs. 5c–d, uncultured_bacterium_f_ Acidimicrobiaceae and Erythrobacter were identified as keystone taxa in microbial communities at domestication stage and operation stage, respectively. Generally, keystone taxa play vital roles in keeping the structural and functional stability of the microbial community under varied environmental conditions [48]. Nevertheless, in the present study, keystone taxa occupied low proportions in the microcosms with relative abundances of 0.07%–0.39% and 0.006%–0.50%, indicating that the proportions of genera were not well correlated with their status in the fungal-bacterial networks.
3.4. Equalization Analysis Between Fungal and Bacterial Community Structures
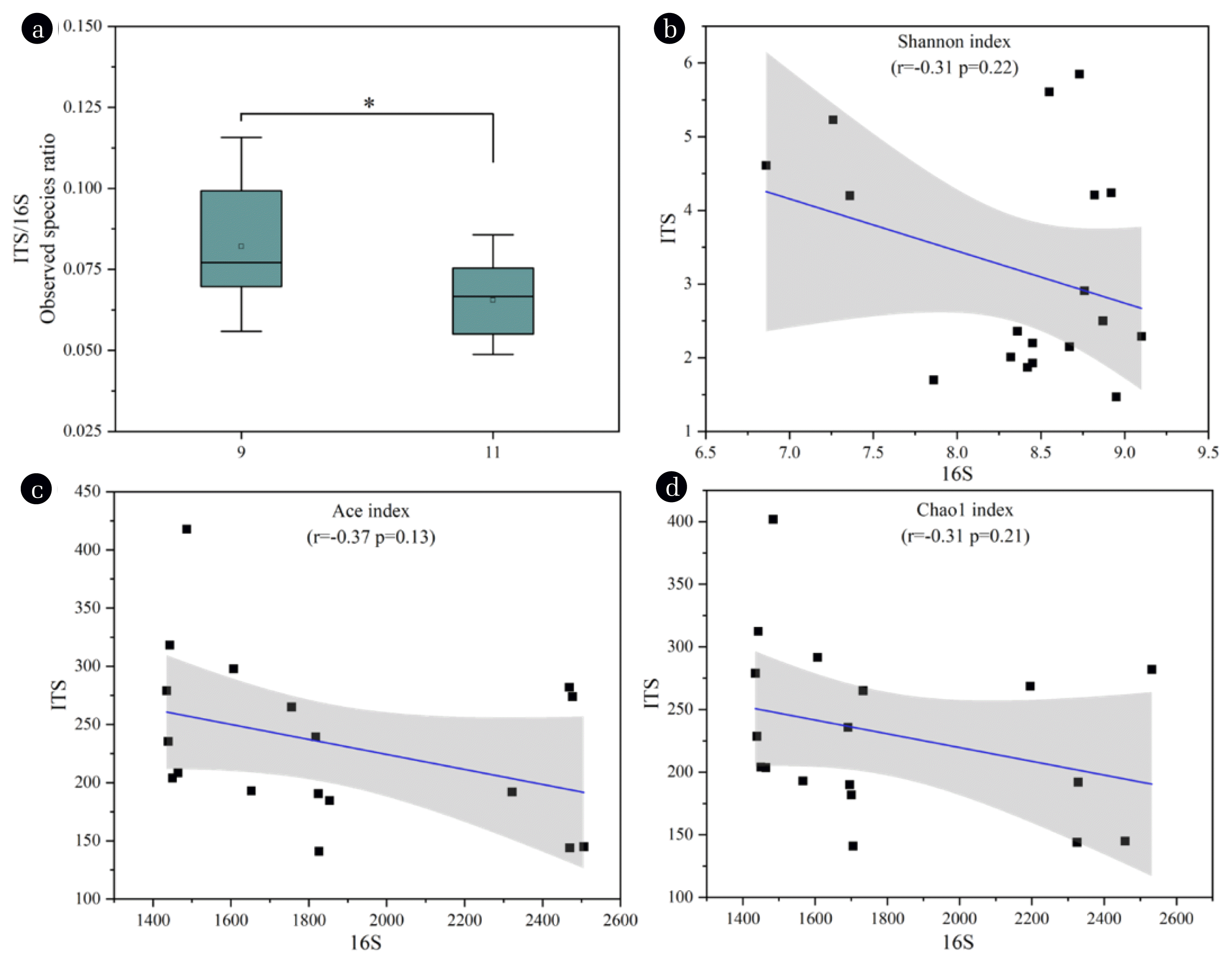
The community equalization is essential for microcosms to maintain the stability [49]. Therefore, an equalization analysis between fungal and bacterial communities was performed. As shown in Fig. 6a, the ITS/16S ratio decreased significantly (P<0.05) at operation stage, indicating a reduction in microbial equalization during this process [50]. The result suggested toxic compounds in the produced water were more likely to exterminate the sensitive fungal populations in the present study. The correlation between fungal and bacterial diversities was examined by Pearson’s correlation analysis, and negative correlations between the alpha diversity indices including Shannon (r=−0.13, P=0.22), Ace (r=−0.37, P=0.13), and Chao1 (r=−0.31, P=0.21) of the microbial communities were found (Figs. 6b–d). This showed that the microbial equilibrium of the bacterial and fungal communities was regulated by the species richness and diversity [50]. Moreover, it could be speculated that a weak competition between fungal and bacterial populations existed in the community, which was detrimental for pollutants decontamination. To improve the wastewater treatment performance, further research is needed to equalize the fungi-bacteria system in the MBCOR.
4. Conclusion
This study investigated the fungal community succession and microbial interactions in the MBCOR during the treatment of low-COD and high-salinity oilfield produced water. Results showed that COD was the main environmental factor in shaping the fungal community within the first tank. Besides, LEfSe analysis suggested that the community structure succession was driven by functional requirements. Additionally, network analysis revealed a strong synergistical cooperation between fungal and bacterial communities, which is in favor of the wastewater treatment. Moreover, variations in species richness and diversity were the principal cause for the change in fungal-bacterial equilibrium in the MBCOR. The findings obtained in this study give deeper insights into the microbial mechanisms during the oily wastewater treatment, providing an instructive guidance for improving the treatment performance of the MBCOR.