1. Introduction
Benzo[a]pyrene, a polycyclic aromatic hydrocarbon (PAH), is considered to be carcinogenic [1, 2]. It is classified by the United States Environmental Protection Agency (US EPA) as a class B2 carcinogenic compound with a mutagenic mode of action for inducing tumors [1, 2]. Additionally, the compound is regulated under the Korea Land Conservation Act of 2009. As a result, the interest in the biotic and abiotic processes for the destruction of hazardous materials in contaminated soil is renewed.
Creosote oil has been used for the preservation of railroad timbers for over 130 years in North America. Pentachlorophenol (PCP) and creosote oil were widely used before 1900 for railroad timbers, electric power poles, and bomb wood boxes [3, 4]. In Korea, waste timbers from railroads are usually recycled as landscaping material after 10–15 years of use [4]. Between 2005 and 2007, about 250,000 pieces of waste railroad timbers were recycled for use in playgrounds, trails, and timber houses. Kim et al. [5] reported that PAHs are major components of creosote oil (about 75%–80%). Therefore, it is expected that the soil around the recycled waste timbers would be heavily contaminated with PAHs [4–7]. Chung et al. [4] also reported that waste railroad timbers contain high concentrations of PAHs, PCP, and benzene, toluene, ethylbenzene, and xylenes (BTEX). Sixteen PAH compounds were tested in waste timbers, and the maximum total PAH concentration was found to be 23,607 mg/kg, including a benzo[a]pyrene concentration of 153 mg/kg. These xenobiotic compounds are known to be toxic to microorganisms and carcinogenic to humans.
Barr and Aust [8] reported that lignin-degrading white fungi are able to degrade lignin as well as hazardous organic pollutants through the peroxidase catalytic cycle, which includes the protein heme. Heme, hemoglobin, and peroxidases (i.e., lignin peroxidase and horseradish peroxidase) react with hydrogen peroxide (H2O2) to degrade PAHs, PCP, crystal violet, and dibenzothiophene [7–11]. We were interested in the development of a soil-remediation technology based on the catalytic/degrading abilities of heme and H2O2 [7, 11]. Our previous work has demonstrated that PCP-contaminated soil was significantly remediated in the presence of heme and H2O2, possibly through a postulated catalytic reaction, under both laboratory conditions and at a pole yard field in Vancouver, WA, USA [11]. We have shown that about 80% of PCP was degraded within 4 hr in the presence of 8.5 g of heme and 47.5 g of H2O2/kg soil.
The objectives of the present work were to demonstrate the heme catalytic mechanism using 5-aminosalicylic acid (5-ASA) as a model organic chemical and to prove that PAHs in soil can be degraded by the catalytic mechanism, including heme and H2O2. We assessed the interaction between heme (Hm-Fe+3) and H2O2 in the presence of 5-ASA and the degradation of PAHs in contaminated soil under an optimized dose of heme and H2O2.
2. Materials and Methods
2.1. Chemicals
Hemin, 5-ASA, 30% H2O2, and trichloroacetic acid (TCA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Reagent-grade chemicals for phosphate buffering were purchased from Duksan Pharmaceutical Co. (Ansan, Korea). The heme stock solution (388 μM) was prepared just before each experiment by dissolving 2.53 mg of hemin in 1 mL of 100-mM NaOH with gentle mixing, immediately adding 9 mL of 50-mM phosphate buffer (pH 7.0) and vortexing. The H2O2 solution was prepared to the desired concentration by diluting 30% H2O2 before each experiment. Five percent TCA was prepared and added to the reaction mixture for the precipitation of heme.
2.2. Spectral Characterization of Heme Catalytic Reactions
Spectra and kinetic measurements for the heme catalytic reaction were obtained using a UV/Vis recording spectrophotometer (UV-1650PC; Shimadzu, Tokyo, Japan). All catalytic reaction mixtures contained 60-μM heme, 50-mM phosphate buffer, 100-μM 5-ASA, and 600-μM H2O2 in a 3-mL total volume using a 5-mL 6Q Quartz cuvette (light path 10 mm; Starna Scientific Ltd., Essex, UK) at 24°C ± 2°C. The catalytic reactions were initiated by the addition of 20 μL of H2O2 (600 μM). The oxidation and reduction states of heme were determined by scanning between 250 and 700 nm. To study the heme catalytic mechanism, we assessed the ability of heme to catalyze the H2O2-dependent oxidation of 5-ASA. The catalytic reactions were terminated by adding 1 mL of a solution containing 5% (w/v) TCA and vortexing for 10 sec. The coagulated heme was then removed from the reaction mixture by filtration using a 0.2-μm filter (Acrodose LC 13 PVDF; Gelman Sciences Inc., Ann Arbor, MI, USA) and scanned in a 1-mL QS Quartz cuvette (Hellma Analytics, Mullheim, Germany).
2.3. Degradation of PAHs in Soil by Heme Catalytic Reactions
The pan study was performed using soil from a pole yard in Vancouver, WA, USA. Five hundred grams of pole yard soil was placed in a stainless steel pan. First, 10 g of heme was dissolved with 40 mL of 0.05-M NaOH and diluted to 400 mL of the final volume with 50-mM phosphate buffer (pH 7.0) to make the feed solution. The heme feed solution was mixed with soil contaminated with PAHs; then, 54 g of H2O2/kg soil was added and mixed. The PAH samples were extracted using a Soxhlet apparatus (US EPA Method 3540A), and the solvent extract was prepared for HPLC analyses using an acid-base partition cleanup (US EPA Method 3650). The solvent extracts were analyzed using HPLC (SIL-6B, Shimadzu) with a UV detector (LC 90 UV spectrophotometric; PerkinElmer, Waltham, MA, USA).
3. Results and Discussion
3.1. Oxidation of 5-ASA by Heme Catalytic Reaction
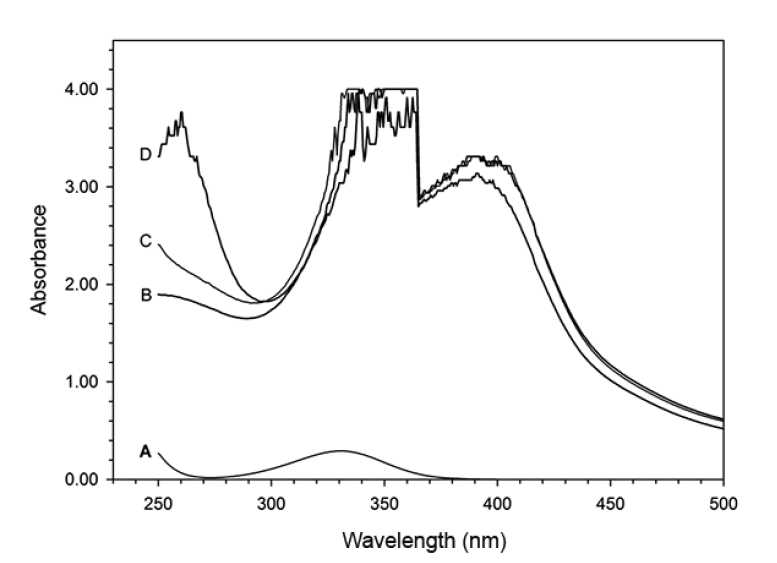
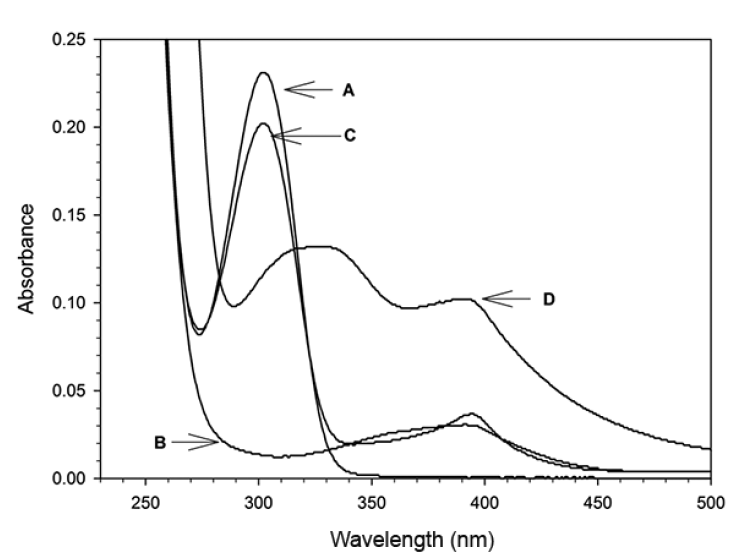
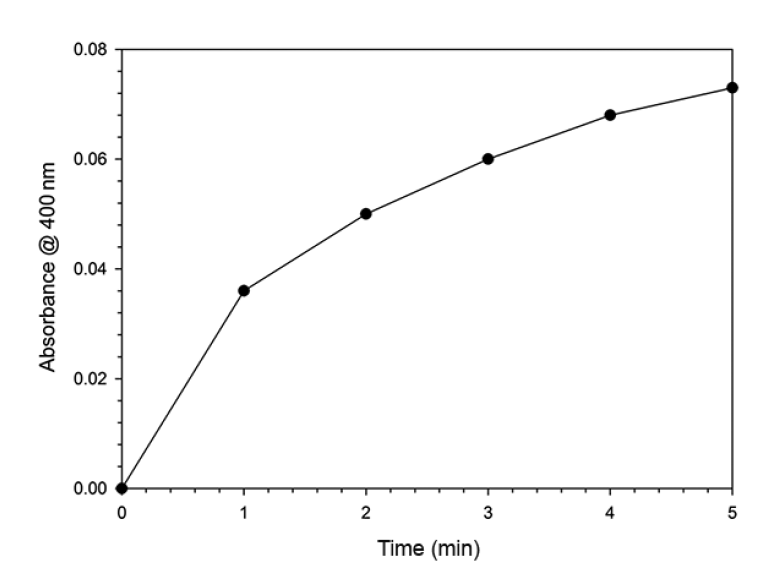
UV/Vis spectra were obtained to study the interaction of heme with H2O2 in the presence of 5-ASA (Fig. 1). All catalytic reaction mixtures contained 60-μM heme, 50-mM phosphate buffer (pH 7.0), 100-μM 5-ASA, and 600-μM H2O2 in a total volume of 3 mL. The spectral characteristics were scanned between 250 and 700 nm. However, the maximum absorbance of heme (i.e., the Soret band) was not clearly observed because of the interference from the other reagents, such as H2O2 and 5-ASA. Thus, the experiments in Fig. 1 were repeated after the heme was precipitated by the addition of 1 mL of 5% TCA and removed by filtration [6]. Fig. 2 illustrates the UV/Vis absorption spectra for the mixtures of 5-ASA, heme, or H2O2 at pH 7.0. The absorption spectrum did not significantly change in the presence of either 5-ASA or 5-ASA/heme (Fig. 2(a) and (c)). In the presence of heme and H2O2 without 5-ASA, the peak at 300 nm was not observed and a weak peak appeared at 400 nm (curve B in Fig. 2). These results indicate that the peak at 300 nm can be attributed to 5-ASA while the weak peak at 400 nm is due to heme. In the spectrum for the mixture of all three reactants (heme, H2O2, and 5-ASA), strong absorption peaks at 330 and 400 nm were observed, possibly because of the formation of oxidized 5-ASA (5-ASAox) by the heme catalytic oxidation reaction (curve D in Fig. 2). Thus, 5-ASA was oxidized only in the presence of both heme and H2O2. It is known that 5-ASAox has an absorption peak at 400 nm that can be used to measure the change in its concentration [12, 13]. The formation of 5-ASAox was confirmed by measuring the change in absorbance at 400 nm. The oxidation of 5-ASA in the presence of heme was initiated by the addition of H2O2. The reaction was stopped by the addition of 1 mL of 5% TCA solution and the precipitate was filtered to measure the absorbance at 400 nm. Fig. 3 shows that the absorbance at 400 nm increased over time and therefore that 5-ASA was oxidized by heme catalytic reactions.
3.2. Heme Catalytic Mechanism
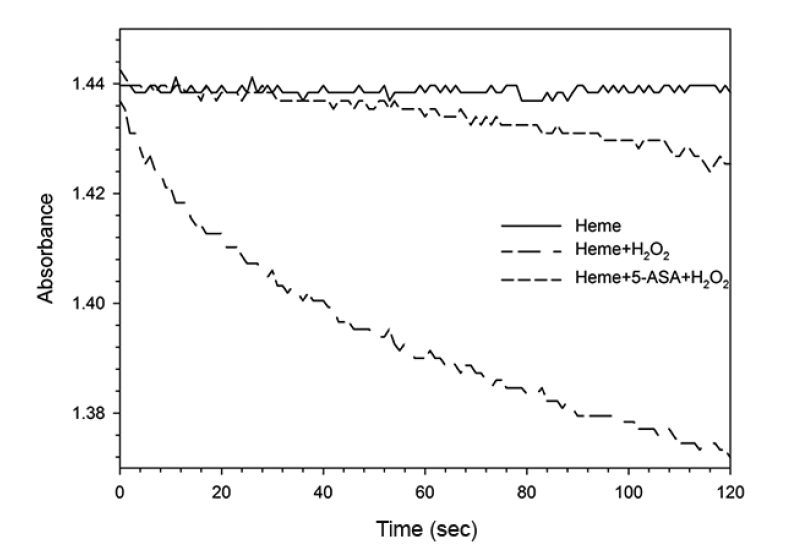
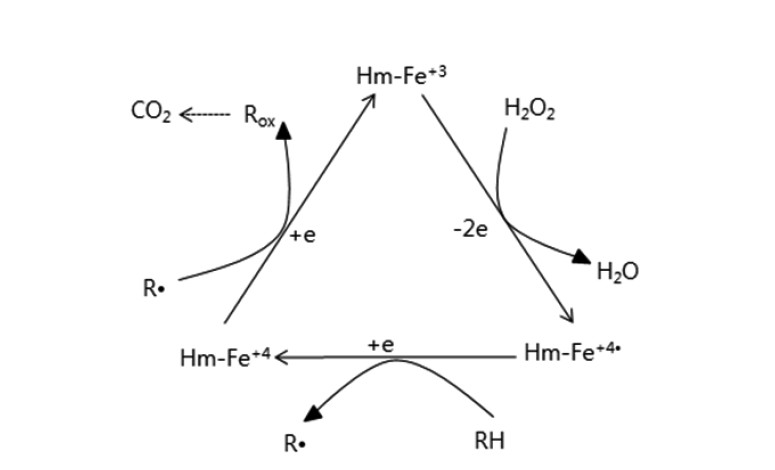
The mechanism of the heme catalytic reaction has been postulated to be similar to that of peroxidase or hemoglobin [10, 13]. However, no clear experimental data exists to explain the mechanism. In our experiments, heme in the phosphate buffer (pH 7.0) had a maximum absorbance at 360 nm (Fig. 4, peak A). When H2O2 was added to the solution containing heme, the absorbance decreased from 1.450 to 1.191 (Fig 4, peaks A and B). This reaction suggests that ferric heme (Hm-Fe+3) was oxidized to ferryl heme (Hm-Fe+4) through the ferryl heme radical (Hm-Fe+4•) upon reaction with H2O2 [14]. When 5-ASA was added to the reaction mixture, the absorbance increased from 1.191 to 1.307. This result suggests that the ferryl heme radical (Hm-Fe+4•) oxidized 5-ASA to 5-ASA radical and was reduced back to ferric heme (Hm-Fe+3) with one more oxidation of 5-ASA or 5-ASA radical to complete the catalytic or redox cycle (Fig. 4, peak C), while 5-ASA as a substrate may be oxidized into 5-ASAox. In support of the data in Fig. 4, the duration of time for the three reactions was 2 min (Fig. 5). These results show that 5-ASA reacted with oxidized ferryl heme radicals or ferryl heme to reduce the oxidized hemes back to ferric heme. The heme catalytic reaction mechanism may therefore be defined as shown in Fig. 6, which is analogous to the catalytic cycle for the degradation of PCP [6, 7, 11, 13].
3.3. Degradation of PAHs in Soil
The oxidized heme intermediate ferryl heme radical (Hm-Fe+4•) is formed by a two-electron oxidation upon interaction between H2O2 and heme. A one-electron oxidation results in the formation of Fe+4 from Fe+3 and another one-electron oxidation is due to the oxidation of the heme porphyrin ring. It has been suggested that the ferryl heme radical (Hm-Fe+4•) is short-lived and that it rapidly decomposes to ferric heme upon reaction with a substrate by two successive one electron oxidations. A few studies have shown that heme is capable of oxidizing a wide variety of substrates [6, 7, 11, 13, 15, 16]. Specifically, Chen et al. [7, 11] reported significant degradation of PCP in soil in the presence of heme and H2O2. To extend the use of the heme catalytic reaction, we investigated the degradation of PAHs, which are commonly found in soil around wood preserved with creosote oil. The PAH-contaminated soil came from a pole yard in Washington, USA. This contaminated soil was a sandy loam and contained 464 mg of PAH/kg soil. Five hundred grams of the PAH-contaminated soil was placed into a stainless pan and mixed with 400 mL of heme feed solution containing 10 g of heme as described in the Materials and Methods section. Finally, 159 mL of 30% H2O2 was diluted with distilled water to make a final volume of 250 mL and mixed well by spraying into the soil in the pan. The water content in this soil was maintained at 50% for 42 days. The PAH concentrations in the soil decreased from 464 to 263 mg/kg of soil at day 14 and to 10 mg/kg of soil at day 42, as shown in Table 1. Thus, the extent of the PAH degradation was about 96% by day 42. Approximately 40% of the toxic components of the PAHs (e.g., benzo[a] pyrene) were no longer detectable by day 42. Therefore, with the combination of heme and H2O2, PAHs in contaminated soil can be effectively and economically remediated. This technology can be considered to be an innovative technology. We believe that the remediated soil is environmentally safe because heme cannot perform any catalytic activity in the absence of H2O2.
4. Conclusions
The present study focused on the heme catalytic reaction in the presence of H2O2 using substrates, such as 5-ASA. The study also evaluated the practical use of the catalytic reaction in degrading the PAHs present in the soil obtained from a pole yard. We concluded that the reactions in the presence of heme and H2O2 can be explained as a possible catalytic cycle that include ferric heme (Hm-Fe+3), ferryl heme radical (Hm-Fe+4•), and fer-ryl heme (Hm-Fe+4). Even though PAH compounds have been shown to be toxic, carcinogenic, and recalcitrant to microbial degradation, about 96% of the PAH content in the pole yard soil was degraded by heme and H2O2. Moreover, benzo[a]pyrene and six other PAH compounds, out of 16 compounds tested, were undetectable in the soil at day 42. In conclusion, because of its environmental safety the heme catalytic degradation of PAHs can be used as a novel technology for the remediation of hazardous organic compounds.