







1. Introduction
As the consumption of aquatic products increases, the aquaculture industry has continuously grown and consequently, the use of hormones and fungicides is increasing to mass-produce aquatic products, promote growth, control fungus and inhibit ectoparasites in fish eggs, fingerlings, and adult fish [1, 2]. However, residual amounts of fungicides and hormones might be discharged into the water environment after excretion of these organisms [3]. Many studies have reported that such indiscriminate use of hormones and fungicides and even low levels (ng/L) may cause genotoxicity and variability, carcinogenicity, mutagenicity, and teratogenicity in many animal species and cell lines, reduced egg production in fish, and have negative effects on adult development [1, 3–6]. Diethylstilbestrol (DES) is an artificial industrial chemical, widely used as a hormone substitute, oral contraceptive, and growth promoter in livestock breeding since 1938 [7]. However, this compound was banned in some countries as it was found to have adverse effects on humans and animals [8]. Medroxyprogesterone acetate (MPA), on the other hand, is the most widely used hormone among various types of synthetic progestins [9–14]. Research showed that MPA affects adult development of fish and decreases egg production even at environmentally similar concentrations [5, 6, 15, 16]. Malachite green (MG) and gentian violet (GV) are representative prohibited fungicides in aquaculture due to their high effectiveness. Their metabolites, leuco-malachite green (LMG) and leuco-gentian violet (LGV) have been reported to persist in the tissues of exposed fish with LMG having a longer half-life than MG [17–20]. Studies have shown that GV and LGV are potential carcinogens, known to cause malformations and mutations [21–26]. MG and GV are structurally related to other triphenylmethane dyes such as rosaniline, which has been linked to an increased risk of human bladder cancer. The leuco form of rosaniline induces renal, hepatic, and lung tumors in mice [17]. Studies show that LMG is an in vivo mutagen and the mutagenicity of MG and LMG correlates with their tumorgenicities in mice. Compared to LMG, MG is much less toxic [17, 19]. DES is currently listed in Group 1 as a carcinogen in the IARC classification list while MPA is listed in Group 2 as a possible carcinogen [7].
Although the use of these chemicals has either been banned or restricted to ‘prohibited substances’, these compounds are still being used illegally in some areas because of their low cost and high efficacy [27, 28]. The concern about the adverse effects of these compounds drives the need for sensitive and selective analytical methods for the determination of these compounds in real samples such as seafood and water samples. Various analytical methods of hormones or fungicides in biota samples have been developed including capillary electrophoresis [29], immunoassay method [30], liquid chromatography with amperometric detection [31]. Furthermore, pretreatment methods for fungicides were mostly performed by a single analysis method for each group [32, 33]. In studies related to the analysis of hormones and fungicides, techniques as offline solid phase extraction (SPE) [28, 34–38] or online SPE [13], liquid-liquid extraction (LLE) [2], solid-phase microextraction (SPME) [39], molecular imprinting technique coupled with SPE (MISPE) [27] have been previously reported for the pretreatment of parent compounds and/or metabolites in overseas water environments. There however, still remains very limited studies reporting the analytical methods and simultaneous pretreatment of these target compounds in seawater samples [27, 40–42].
Therefore, in this study, a simultaneous analytical method for fungicides and their metabolites (MG, GV, LMG, and LGV) was developed, in addition, a simultaneous analytical method for hormones (DES and MPA) was developed using LC-MS/MS. The developed analytical methods were applied to real seawater samples for method validation. This is the first study to provide a concise and effective pretreatment method that allows simultaneous analysis of these prohibited substances and their metabolites in seawater samples.
2. Materials and Methods
2.1. Chemicals and Standards
Two target hormone standards comprising of DES, MPA, and four fungicide standards comprising of MG and LMG, GV and LGV were purchased from Sigma-Aldrich (St Louis, MO, USA), Dr. Ehrenstorfer (Augsburg, Germany), and Cambridge Isotope Laboratories (Andover, MA, USA). The physicochemical properties of these compounds are shown in Table S1. Five internal standards (DES-d8, MP-d3, LMG-d6, GV-d6, and LGV-d6) were purchased from Cambridge Isotope Laboratories Inc. (Tewksbury, MA, United States), Toronto Research Chemical Inc. (Toronto, Ontario, Canada), Dr. Ehrenstorfer, Santa Cruz Biotechnology, and Dr. Ehrenstorfer, respectively, and diluted into one single mixture. The minimum purity of all commercial standards was 95%. Stock standards solutions were prepared at 1000 mg/L with methanol (MeOH) for hormone compounds and acetonitrile (ACN) for fungicides. All chemical reagents were of analytical grade. MeOH, ethyl acetate (EtAC), ACN and ultra-pure water were purchased from B & J Honeywell (Morristown, NJ, USA). Formic acid, ethylenediaminetetraacetic acid (EDTA), hydroxylamine hydrochloride, p-toluenesulfonic acid, ammonium acetate, ammonia, ascorbic acid and hydrochloric acid were each purchased from Wako (Osaka, Japan), Sigma-Aldrich (St Louis, MO, USA), Junsei (Tokyo, Japan) and Matsunoen chemicals Ltd (Osaka, Japan). All organic solvents used for experiments including extraction and purification were of HPLC grade. Oasis HLB (500 mg, 6 cc; Waters, Milford, MA, USA), Oasis HLB (200 mg, 6 cc; Waters), Oasis MCX (150 mg, 6 cc; Waters) cartridges were used for sample extraction and purification. All solutions were stored at −20°C.
2.2. Instrumental Analysis
2.2.1. Fungicides (MG, GV, LMG, LMV) and hormones (DES, MPA)
The chromatographic separation was performed using a high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) (Agilent 1200/6460 triple Quad; Agilent Technologies, Santa Clara, CA, USA). The analytes were separated on a ZORBAX Eclipse XDB-C18 column (2.1 mm × 150 mm × 3.5 μm, Agilent, USA). For fungicides, 0.5% formic acid in ACN and 10mM ammonium acetate and 0.5% formic acid in water were used as the mobile phases A and B respectively. While for hormones, MeOH and 2mM ammonium fluoride in water were adopted as the mobile phases A and B respectively. For quantitative analysis, a method of electron spray ionization tandem MS (ESI-MS/MS) was developed. A precursor ion (PI), quantification/qualification ions (QI 1/QI 2) were selected for each target compound and were analyzed by multiple reaction monitoring (MRM). For fungicides, analysis was done in positive mode. For hormones, DES was analyzed in the negative ion mode, while MPA was analyzed in the positive ion mode. Details of the full MRMs and instrumental conditions for fungicides and hormones are shown in Table 1.
2.3. Sample Preparation and Extraction Method
To develop an efficient pretreatment SPE method for all target compounds, the recovery test experiments were performed using several cartridge types, different pH levels, various elution solvents and also different additives. The effect of pH and additives are critical parameters that affect extraction efficiency [41].
2.4. Recovery Rate, Accuracy, and Precision
To establish an optimal pretreatment method, internal standards (DES-d8, MP-d3, LMG-d6, GV-d6, and LGV-d6) were added to seawater samples and each pretreatment method was repeated seven times before LC-MS/MS analysis for repeatability. To calculate the accuracy, the ratio of the measured values to the true values was expressed as a percentage. To calculate the precision, the relative standard deviation (RSD) of the sample injected with the standard was calculated and divided by the average concentration of the target substance and expressed as a percentage. Method detection limit (MDL) was calculated by multiplying the standard deviation obtained through seven repetitions of sample by the degree of freedom (t = 3.143, student’s t-value in 99% confidence interval) as shown in Eq. (1) and the limit of quantification (LOQ) was calculated by multiplying the obtained standard deviation by 10 as outlined in the US EPA (40 CFR Part 136)
-
t(n − 1, 1 − ∝ = 0.99) Student's t value for the single tailed 99th
percentile 0.99 level for n-1degrees of freedom; and -
Ss sample standard deviation of replicate spiked
sample analyses relativestandard deviation
The calibration curve was prepared using a mixed standard solution prepared with a concentration range of six levels (0.2 – 50 ng/mL). The correlation coefficient of the calibration curve (R2) was good for all compounds at 0.99 or higher and was quantified by applying the reciprocal (1/x) of the reference amount. Before the instrument analysis of the hormones, MeOH was injected into the instrument, while before analysis of fungicides, ACN was injected into the instrument to check for any contamination, and we observed there was no background contamination in the method blank.
To check the accuracy of the instrument, the standard solution (20 ng/mL) which was the middle concentration range of the calibration curve was reanalyzed in the final position to check whether the calibration curve changed during the instrumental analysis. Variations of the relative ion intensities were within 20%.
3. Results and Discussion
3.1. LC-MS/MS Condition
At optimum conditions, the precursor and product ions for both hormones and fungicides are shown in Table 1. For DES, optimum conditions were obtained in ESI negative mode while optimum conditions for MPA were obtained in ESI positive mode. Mobile phases, MeOH and 2mM ammonium fluoride were selected for optimal separation. For fungicides, optimum conditions were obtained in ESI positive mode and mobile phases; 0.5% formic acid in ACN and 10 mM ammonium acetate with 0.5% formic acid, were selected for efficient separation. Representative chromatograms for the target compounds in seawater samples are illustrated in Fig. S1.
3.2. Fungicides (MG, GV and their metabolites) Pretreatment Method Optimization
3.2.1. Preliminary approach
For the simultaneous extraction of two fungicide parent compounds and their metabolites, preliminary tests were carried out with the HLB and MCX cartridges which had been found to achieve good recoveries in seawater, natural water samples [41, 43]. Fungicide standards (0.2 ng) were spiked in 1 L of ultrapure water samples and treated with HLB and MCX respectively in various conditions. These methods were adapted and modified from previous studies to assess the pH and additives effect [41, 44] and the detailed method and recovery rates can be seen in Table S2 and Table S3. For methods 1-1 and 2-1, where there was no pH adjustment or use of additives, extremely poor recovery rates of internal standards were observed for all compounds for both the HLB and MCX cartridges ranging from 1.0–58.2% and 8–12.8% respectively. According to [45], pH plays an important role in the extraction procedure because it determines the existing form of the analytes and affects the extraction efficiency. MG occurs in two ionic forms and has two amino groups with pKa of 6.9 that are protonated at pH 4. At pH 10, most molecules are deprotonated and therefore colorless. From the study by Mitrowska K et al. [46], pH levels from 3.0 to 8.0 were tested and the best recovery rates were observed at pH 3.0. In acidic conditions, the carbinol form of MG is converted to the chromatic, cationic form while alkaline conditions increase the possible hydrolysis of LMG and hence decreases its recovery. Formic acid was added to adjust the pH of the sample, thereby increasing the extraction efficiency of the cartridge. In methods 1-2 and 2-2, with the addition of additives and pH adjustment of 3, excessively high recovery rates were obtained ranging from 82.3–251.7% for the HLB cartridge, with improved high recoveries, ranging from 44.6–69.9% for the MCX cartridge. Previous studies showed that additives such as hydroxylamine hydrochloride acted as an antioxidant/reduction inhibitor for LMG and LGV, and p-toluenesulfonic acid was used as an ion pair reagent that formed ion pairs with the analytes, hence they were easily extracted by organic solvent [47, 48]. Overall, in terms of internal standard recoveries, the MCX cartridge (Table S3) were better than the HLB cartridge despite the low accuracy of LGV in MCX.
3.2.2. Optimization for SPE cartridge selection and additives
From previous studies, the ammonium acetate buffer, was shown to have a significant effect on peak resolution and intensity of target compounds [46, 47, 49]. Hence, a buffer test was done on the MCX cartridge, a bar chart of the results is presented in Fig. 1. The buffer test showed significantly better peak responses, with increases of over 100%, for samples to which the ammonium acetate buffer was added.
Furthermore, with the addition of the ammonium acetate buffer (1M ammonium acetate), combined HLB and MCX cartridges (method 3-1), adapted and modified from [38, 50], were tested and recoveries were compared with the MCX cartridge. The cartridges were arranged in tandem with the MCX cartridge above the HLB cartridge. For the combined cartridges, recoveries ranged from 56.1–277.2% with GV still having an excessively high recovery rate (277.2%). Meanwhile, recoveries for all target compounds using the MCX cartridge (method 3-2), ranged from 85.1–140.0% (Table 2). Therefore, method 3-2 was selected as our optimized pretreatment method for the fungicides.
3.2.3. Validation of the method effectiveness of four types of fungicides (MG, GV, and their metabolites) in seawater samples
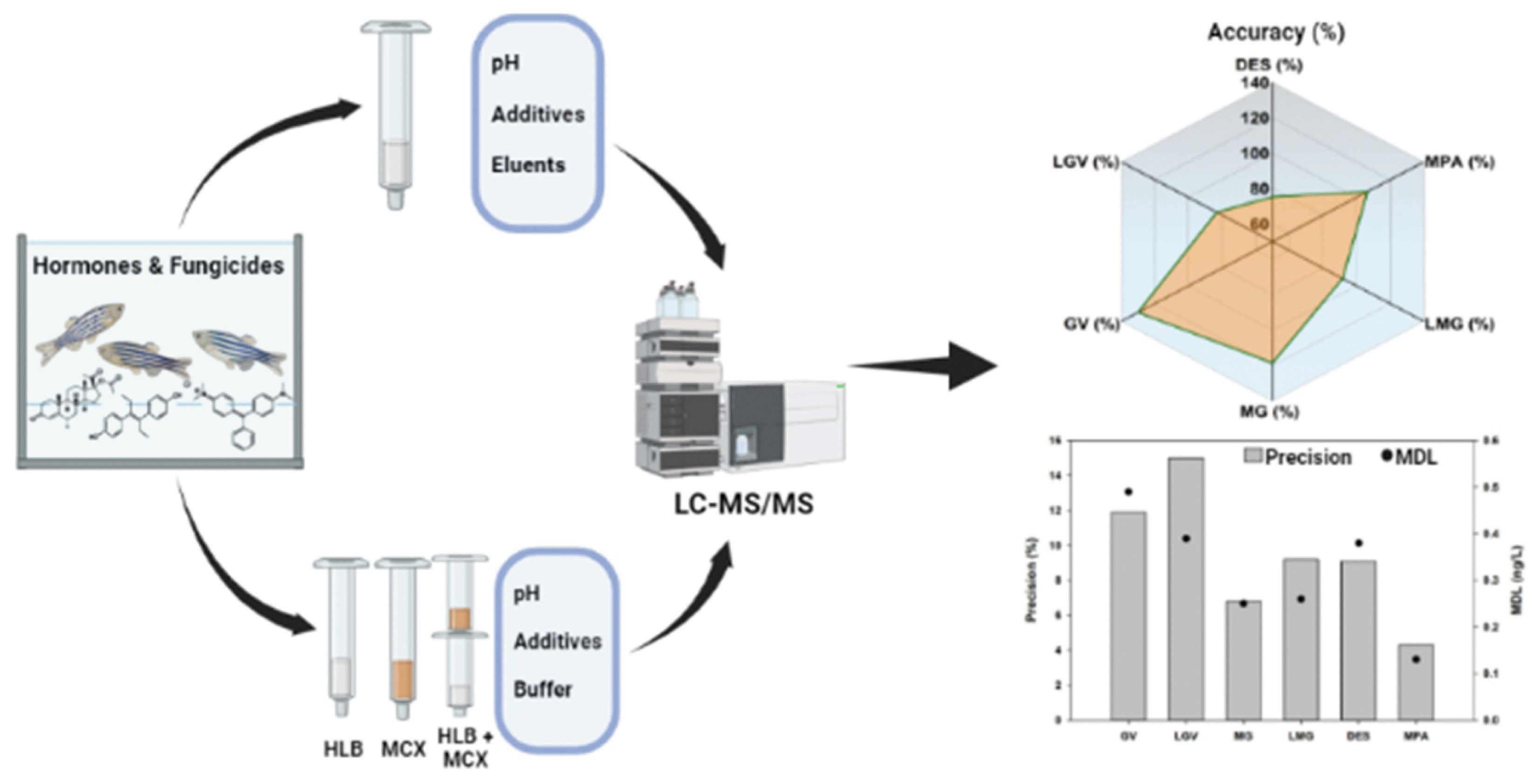
In this study, method validation was performed by determining the recovery, precision, method detection limit (MDL) and limit of quantification using method 3-2. The precision, repeatability and recoveries were calculated for seawater samples spiked with native compounds. Seven replicate analyses were performed using the MCX method (Method 3-2) described in Table 2, with 10 ng of target compounds spiked in 200 mL of seawater samples. Quantitative recoveries of used label standards were in the range 86–90.7%. Target compounds were confirmed with an accuracy of 83.2–130.1%, a precision of 6.8–15.0% at the corresponding concentrations (Table S4). This pretreatment method was therefore considered as an appropriate method for simultaneous analysis. The IDL ranged from 0.003–0.0019 ng/mL, MDLs and LOQs each ranged from 0.25–0.49 ng/L and 0.81–1.55 ng/L respectively (Table 3). Compared to MDLs (0.023 – 4.8 ng/mL) and LOQs (0.35 – 5 ng/mL) in previous studies [27], [41], [51], the target compounds in this study showed better MDL and LOQ values. The results from our study were equally found to be less than 1/10 of the LOQ for these fungicides, 2.0 ng/mL, according to the Korean Food Standards Codex. These results indicate that the proposed method in this study is capable of analyzing trace levels of the fungicide target compounds.
3.3. Hormones (DES, MPA) Pretreatment Method Optimization
3.3.1. Elution solvent test
To check the simultaneous extraction of two hormone compounds, DES and MPA, recoveries for the different elution solvents were compared. In most of the previous studies, the Oasis HLB cartridge (500 mg, 6 mL) was used for pretreatment [28, 34–38]. Elution solvents such as acetonitrile, ethyl acetate, methanol, and dichloromethane were most widely used [13, 28, 34, 37]. In addition, using the HLB cartridge and LC-MS/MS, recovery rates of 87.1–108% were observed for DES and 70–113% for MPA were reported. Studies which used the Atlantic C18 cartridge for MPA showed recovery rates of 58–116% [9]. Therefore, the HLB cartridge was selected for simultaneous pretreatment of hormones. 5 ng of DES internal standard and 20 ng of MP internal standard were spiked in 1 L of ultrapure water, and 12 mL of different elution solvents were used (Table 4). EtAc:ACN, and EtAc and MeOH methods were first tested (Methods I to III) and then a MeOH method with pH adjustments and additives was further tested (Method III-a).
The recoveries varied according to the different elution solvents used. Table 4 shows the accuracies and recoveries of both compounds in the elution solvent test. In case of EtAc:ACN method (Method I), the average recovery was 1.3% for DES and 67.2% for MPA. For the EtAc method (Method II), the average recovery of DES was 1.2% and that of MPA was 54.4%. Similar to method I, a low recovery was observed for DES. For the MeOH method (Method III), the average recovery of DES was 38.8% and MPA was 63.3%. A relatively high recovery was observed for hormones using MeOH method [3]. According to previous studies, MeOH was shown to be the most suitable when comparing the recoveries based on the choice of extraction solvent for DES compound. However, to compensate for the difference in the expected recovery due to the deviation of the pKa value of DES reported in literature (5.07–9.02), and since the recovery obtained for DES with the MeOH method was low, a pH adjusted MeOH method with additive was further tested (Method III-a). As previously stated, for the fungicides, in solid phase extraction, pH conditions and additives play a very significant role in improving the extraction efficiencies of the cartridge. Here, the sample pH was adjusted to 2.5 with HCl, in consideration of the lowest pKa value, and oxidative decomposition of the phenolic OH group of DES was inhibited by addition of L(+)-ascorbic acid [52]. The resulting average recovery of DES was 67.3% and MPA was 117.4%. From this study, the pH and additive played a very important role in the extraction efficiency. Previous studies also showed similar results, where adjusting the pH and changing the eluting solution produced optimal recoveries. After testing MeOH, acetone, and ACN, MeOH was selected as eluent due to its highest recoveries and lower toxicity [3].
3.3.2. Validation of the hormones (DES, MPA) pretreatment method in seawater samples
In this study, to evaluate the validity of the pretreatment method, an analysis was performed to establish the accuracy, precision, recovery rate, MDL, and LOQ of the target compounds in seawater. The pretreatment was performed in the same way as the pH adjusted MeOH method with additive (Method III-a), using seven replicate samples, 10 ng of native standard was added to 200 mL of seawater. The results are presented in Table S5. All hormone target compounds showed accuracies between 75.3–106.6% at the corresponding concentration, and all target compounds showed precision of 4.3 – 9.1% (within 10%). Therefore, when considering the recovery for each pretreatment method, the pH adjusted MeOH method with additive was considered to be the most appropriate. The MDLs obtained in this study were 0.38 ng/L for DES, and 0.13 ng/L for MPA, LOQs were 1.22 ng/L for DES, and 0.40 ng/L for MPA and IDLs were 0.0015 ng/mL for DES, and 0.0084 ng/mL for MPA (Table 5). Comparing our MDLs and precisions, to another previously reported study focused on seawater samples, our MDL for MPA was much less than this study (0.50 ng/L), and similar to that for DES (0.25 ng/L), while our precision values were similar to this study [53] (6.2% for DES and 3.5% for MPA). The results from our study were found to be less than 1/10 of the LOQ for DES 0.2–0.5 ng/mL and MPA 0.8–3.0 ng/mL for livestock and aquatic products according to the Korean Food Standard Codex.
4. Conclusion
The use of fungicides and hormones has increased due to the increase in demand for aquaculture products. Although the use of some of these compounds has been prohibited in some countries due to their toxicity, their low cost and easy accessibility has led some of them to still be illegally used. With no standard method for the simultaneous pretreatment of these compounds in seawater, there is a need to develop and establish an analytical method. Therefore, in this study, pretreatment methods for the simultaneous analysis of 2 fungicides (MG, GV) and their metabolites (LMG and LGV), and 2 hormones (DES, MPA) were established. The pretreatment method using the MCX cartridge was selected as the optimized method for the fungicides, while pH adjusted MeOH was selected as the optimized extraction solvent in the pretreatment method for the hormones. We found that we obtained lower MDL and LOQ values with our pretreatment methods compared to some other previous studies. Also, the MDLs from our above methods were less than 1/10 of the LOQ for livestock and aquatic products. The results of this study can be used as a basis for the efficient and simultaneous analysis of banned hormone and fungicide compounds in seawater samples lacking in research in Korea in the future. It could also be expanded to monitor and preemptively respond to future illegal use of target compounds.



