In situ measurement-based partitioning behavior of perfluoroalkyl acids in the atmosphere
Article information

Abstract
Environmental fate of ionizable organic pollutants such as perfluoroalkyl acids (PFAAs) are of increasing interest but has not been well understood because of uncertain values for parameters related with atmospheric interphase partitioning behavior. In the present study, not only the values for air-water partition coefficient (KAW) and dissociation constant (pKa) of PFAAs were induced by adjusting to in situ measurements of air-water distribution coefficient between vapor phase and rainwater but also gas-particle partition coefficients were also estimated using three-phase partitioning model of ionizable organic pollutants, in situ measurements of PFAAs in aerosol and air vapor phase, and obtained parameter values. The pKa values of PFAAs we obtained were close to the minimum values suggested in literature except for perfluorooctane sulfonic acids, and COSMOtherm-modeled KAW values were assessed to more appropriate among suggested values. When applying parameter values we obtained, it was predicted that air particle-associated fate and transport of PFAAs could be negligible and PFAAs could distribute ubiquitously along the transection from urban to rural region by pH-dependent phase transfer in air. Our study is expected to have some implications in prediction of the environmental redistribution of other ionizable organic compounds.
1. Introduction
Per- and polyfluoroalkyl substances (PFASs) are global pollutants, which were listed as persistent organic pollutants (POPs) by the Stockholm Convention [1]. Once released into the environment, organic pollutants experience various phase-partitioning between solid phases (e.g., soil, sediment, air particle, and biota, etc.), water phase, and air vapor phase. Particularly, atmospheric fate and transport plays an important role to their redistribution among environmental compartments and long-range transport, as frequently mentioned as ‘grasshopper effect’ [2].
In atmosphere, the partitioning behavior of neutral PFASs such as fluorotelomer alcohols and perfluooctane sulfonamidoethanols have been predicted comparatively easily using their two-phase partitioning coefficient (e.g., gas-particle partition) because of their less soluble property [3, 4]. Acid PFAS group (perfluoroalkyl acids (PFAAs)) exist as the free acid form (i.e., neutral species) and their conjugate base form (i.e., ionic species) as shown in perfluoroalkyl carboxylic acids/carboxylates (PFCAs) or perfluoroalkane sulfonic acids/sulfonates (PFSAs) [5]. Differently from neutral PFASs, the environmental fate of PFAAs has been less known and is still in debate, which is related with the presence of both neutral and ionic species and the significant uncertainties in their physicochemical properties such as dissociation constant (i.e., pKa) and air-water partition coefficient (KAW) [6–8].
PFAAs are dissociated into their ionic species when they meet the aqueous phase and in the typical aquatic pH conditions are known to exist as mostly ionic species, which is difficult to be volatilized [8]. Therefore, their airborne transport mechanism has been ignored for long time because PFAAs were considered to be rarely present in the atmosphere. Their presence observed in rain water, had not been well explained, was later attributed to the transformation of volatile and less soluble precursors (i.e., neutral PFASs) to soluble PFCAs and PFSAs [6, 7]. In our previous study [8], however these ionizable organic pollutants (i.e., PFCAs and PFSAs) were measured in rain samples as well as air samples such as gas- and particle-phases. Furthermore, gradually decreasing distribution pattern from source (urban) area to non-source (rural) area could not be explained by previous precursor-based transport mechanism [6, 7, 9]. Furthermore, elevated occurrence of PFAAs in surface runoff samples implicated that their redistribution among environmental compartments through air-water (or surface) partition might occur significantly [8, 10]. Since then, several studies also confirmed the presence of PFAAs in atmospheric compartments including aerosol and air vapor phases [11–13]. As a possible mechanism to account for the occurrence of PFAAs in atmosphere, McMurdo et al. [14] proposed “PFAAs enrichment in aerosol by the splashing of surface water and occurrence of gaseous PFAAs by gradual volatilization of water molecules”. Thus, the redistribution of PFAAs through atmospheric transport can be more significant in coastal region or urban area with large human and/or industrial activities and large water body, considering that 1) surface water can be easily splashed into air by strong wind and wave, 2) their ionic species can exist abundantly in seawater (particularly, they can highly accumulate in the surface microlayer) [15], and additionally 3) fog can frequently be formed owing to difference of air-water temperature [16]. By redistribution process in the environment, human being may be exposed to these toxic substances through multiple pathways via drinking, food intake, dermal contact and inhalation [17, 18].
Differently from insoluble and neutral POPs, ionizable organic pollutants such as PFAAs distribute among three phases in atmosphere, including particle phase, vapor phase, and aqueous phase [11, 12, 14]. The redistribution of PFAAs among environmental compartments thus may be significantly influenced by the atmospheric three-phase distribution (or partitioning) behavior which strongly depends on the extent of their dissociation [19]. It is thus indispensable to use accurate values for the parameters related with their dissociation such as pKa, solution pH, and water volume (e.g., relative humidity) in predicting their atmospheric interphase-partitioning behavior. Until recently, wide ranges of pKa and KAW (~10,000 times difference between minimum and maximum values) have been suggested (see references described in section 3.2). Uncertain parameter values could be significant factors amplifying the uncertainty in the prediction of atmospheric fate and transport, including air-water distribution coefficient (DAW), gas-particle partition coefficient (KP′) dry/wet deposition, and long-range transport potential [12, 20–22].
The present work aimed 1) to estimate the reliable pKa and KAW values by adjusting t to in situ DAW measured between air vapor phase and rainwater, 2) to induce KP using in situ measured gas-aerosol partition coefficient (KP′) and adjusted values for KAW and pKa, and finally 3) to predict their occurrence in each phase of atmosphere with variation of meteorological condition.
2. Materials and Methods
2.1. Sampling and Chemical Analysis
The present study focuses on the interpretation of the partition behavior of ionizable organic compounds such as PFAAs, based on our previous measurements [8]. Thus, only brief information on sampling of atmospheric phases was described here and the more details of sampling and chemical analysis were presented elsewhere [8].
In brief, three atmospheric phases (air gaseous and particulate phases, and rainwater) were collected at the Washington Park in downtown Albany, New York with a population of 96,000. The Washington Park (ca., total area of 360,000 m2) lake has a water surface area of 2 × 105 m2 and an average depth of 3 m (so, lake water volume = ~70,000 m3). All of the atmospheric samples were collected on the roof of a lake house building about ten meters above the surface of lake, located at the Washington Park. Gaseous and particulate phase of air (n = 8) were collected from May to July, 2007 using a high-volume air sampler. PUF/XAD-2/PUF sandwich (Supelco, Bellefonte, PA) and quartz fiber filter (QMA, Whatman) were used to collect gaseous and particulate phases, respectively. The collected air samples were kept at 4°C with prior to analysis and were extracted with no desiccation in a few days after sampling. Desiccation of QMA can cause a removal of water phase on the filter and sequential volatilization of neutral species in water phase. Thus, no desiccation of QMA sample can prevent the potential loss of neutral species. Rainwater samples (n = 3) of ~200 mL were collected in methanol-rinsed polypropylene bottles equipped with a funnel next to air sampler.
2.2. Partition Behavior of PFAAs Among Atmospheric Compartments
Once organic acids such as PFCAs and PFSAs are dissociated in aqueous phase, the acids (hereafter, neutral species) are in equilibrium with their conjugate base (hereafter, ionic species) as shown in an example of perfluorooctanoic acids (PFOA) Eq. (1-1) and (1-2). Such a dissolution yields the presence of both neutral species (C7F15COOH) and ionic species (C7F15COO−) that strongly depends on their dissolution constants (pKa) and pH of aqueous phase as shown in Eq. (1)–(2).
Typically atmosphere consists of aerosol phase (air particle phase and water droplet phase) and air vapor phase. As POPs, well known as representative neutral organic compounds, are rarely water soluble, their KP can be well modeled using a two-phase (i.e., air vapor-particle) sorption equation where a water droplet phase of aerosol can be ignored [23]. However, PFAAs can be present in all of three phases and then their speciation in vapor and water phases should be quantified in advance, being expressed by DAW (dimensionless) incorporating both neutral and ionic species as shown in Eq. (2).
where DAW indicates the ratio of the concentration of neutral species in air vapor to the total concentration of neutral and ionic species in aqueous phase at the given solution pH. The KAW is the air-water partition coefficient of neutral species (m3_water/m3_airvapor).
Gas-particle partitioning can be quantified on basis of the measured concentrations as expressed in Eq. (3), in which the term of CP ′(unit; mol/m3) indicates the measured concentrations of all PFAA species present on aerosol that is a sum of neutral species adsorbed on particle, neutral species in water phase, and ionic species in water phase, and the denominator (CA ; mol/m3) indicates the measured concentration of neutral PFAA species present in vapor phase.
where TSP is the mass concentration of total suspended particulate matter in air (μg/m3_air). Although KP ′ based on the measured CP ′ and CA indicates TSP-normalized partition coefficient, PFAAs on aerosol (CP ′) include both neutral and ionic species in aerosol (i.e., particle-phase and water-phase) and then can vary with the volume of water phase (i.e., relative humidity). Therefore, it is necessary to convert KP ′ to the gas-particle partition coefficient independent on the volume of water-phase (KP ), which is known to be a conventional gas-particle partition coefficient for insoluble organic pollutants.
Contrary to KP ′, the KP is defined as the partitioning coefficient between air vapor and particle for the neutral species alone. Meanwhile, it is difficult to isolate and measure solely neutral species adsorbed on particle-phase because both neutral and ionic species exist together in aerosol consisting of particle and water-phase. Furthermore neutral species can be lost via evaporation when filter paper is desiccated. Alternatively, the KP ′ can be converted to KP by multiplied by the fraction of particle-adsorbed neutral species to total species in aerosol (αneutral on particle ′) as given by the following expression:
Likewise, the fractions of neutral species adsorbed on the air particle (αneutral on particle ), neutral species dissolved in water phase (αneutral in water ), ionic species in water phase (αionic in water ), and neutral species in air vapor (αneutral in air vapor ) in total air which consist of gaseous, particulate, and water phases can be calculated as follows:
where Cneutral on particle, Cneutral in water, Cionic in water, and Cneutral in air vapor are the concentrations of neutral species adsorbed on air particle (mol/μg_air particle), neutral species dissolved in water phase of air (mol/m3_water phase of air), ionic species dissolved in water phase of air (mol/m3_water phase of air), and neutral species in air vapor phase (mol/m3_air vapor), respectively. The VW_rH, and Vair vapor indicate the volume of water phase (m3_water droplet/m3_air) at a given relative humidity (rH) and the volume of air vapor phase (m3_air vapor/m3_air) in an unit air volume (Vair; 1 m3_air), respectively. Strictly speaking total air volume is the sum of the volumes of air vapor (Vair vapor), air water (VW_rH), and air particle (Vair particle), but practically Vair (= 1m3 ) ≅ vair vapor because the faction of VW_rH and Vair particle account for just 10−5 (at relative humidity = 50%) and 10−11 (at a typical TSP = 60 μg/m3), respectively. Dividing Eq. (5-1) to (5-4) by Cneutral in waterVW _rH yields
where KPW is the particle-water partition coefficient (unit; m3/μg) which is frequently used to explain partitioning between sediment and water in aquatic system. The KPW is associated with organic carbon-water partition coefficient (KOC) or octanol-water partition coefficient (KOW) as shown in Eq. (7).
where β is a correction factor (= 1012) to match the units of KPW (m3/μg) and KOC (L/kg). The fOC is the fraction of organic carbon in particle (dimensionless) and 0.2 was used for fOC of air particle [24, 25].
Again, the fractions of individual-phase of neutral and ionic species in aerosol are induced from Eq. (6-1) to (6-4) and can be obtained as follows:
where we used the relationship of KOC = 0.41KOW [26].
3. Results and Discussion
3.1. PFAAs Measured in Atmospheric Compartments
Mean concentrations of PFAAs used for atmospheric partitioning of this study were listed in Table 1. Of six PFCAs, PFOA was the most prevalent compound which was detected in all samples of air vapor (n = 8), air particle (n = 8), and rainwater (n = 3). Perfluoroheptanoic acids (PFHpA), perfluorononanoic acids (PFNA), perfluorodecanoic acids (PFDA) were detected above LOQ in all samples except for only one air particle sample, two air particle samples, and one rainwater sample, respectively. More highly fluorinated PFCAs, perfluoroundecanoic acid (PFUnDA) and perfluorododecanoic acid (PFDoDA), were less frequently detected. Particularly, air particle samples did not contain any detectable PFAS concentration include neutral and ionic species. Values < LOQ was treated to ‘zero’. level of PFUnDA and contained PFDoDA above LOQ in only two samples. This may be attributable to rare occurrence of the highly fluorinated compounds in atmosphere. Otherwise, a relatively high particle-phase concentration would have been observed considering their relatively high octanol-air partition coefficients (KOA) compared with C7–C10 PFCAs.
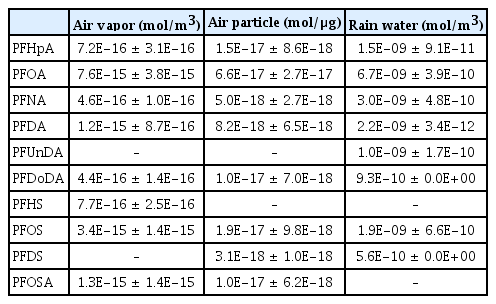
Concentrations of PFAAs Measured in Atmospheric Compartments
Of four PFSAs, perfluorooctanesulfonic acid (PFOS) was also predominantly found in three atmospheric phases. The other PFSAs were not at all detected at least one of three phases; that is, perfluorohexanesulfonic acid (PFHS) in air particles and rainwater, perflorodecanesulfonic acid (PFDS) in air vapors, and perfluorooctane sulfonamide (PFOSA) in rainwaters were not detected. Thus, it was not allowed to calculate their DAW values and to predict their pKa for the three PFSAs.
3.2. Estimation of pKa from Observed DAW
As mentioned earlier, the partitioning of PFAAs among atmospheric compartments strongly depends on their pKa and KAW and solution pH (Eq. (1)–(8)). A pH of 4.5 was used for a typical rainwater and air water phase on the basis of the meteorological data measured at urban area including northeastern states of US and Canada [11, 27]. For pKa, a number of values for PFAAs have been suggested so far, which were estimated theoretically [28, 29], calculated from computational approach using software COSMO-RS [30, 31] and SPARC [32], or measured from laboratory experiment [33–35]. The suggested pKa values ranged from -0.21 to 3.8 for PFOA and from −3.3 to 0.14 for PFOS (Table 2). Various two-phase partition coefficients including KAW, KOW, and KOA have been also estimated from molecular structure (i.e., quantitative structure-activity relationships) using a variety of tools such as U.S. EPA’s EPI Suite, ClogP, SPARC, and COSMOtherm [36]. According to previous results (see the literature in Table 2), those QSAR-based models produced the partition coefficients that differed more than three orders of magnitude: for instance, log KAW = −2.37 (COSMOtherm) to 0.57 (EPI Suite) for PFOA and −2.40 (COSMOtherm) to −0.35 (EPI Suite) for PFOS. Recently, Kim et al. [37] who used molar volume as a molecular descriptor suggested KAW values greater than 10 times than the maximum values suggested by previous QSAR-based models (Table 2).

Physicochemical Properties for Fate of PFAAs Recommended in Literature
Such wide ranges of pKa and KAW (~10,000 times difference between minimum and maximum values) could be significant factors enlarging the uncertainty in the prediction of atmosphere-associated fate and transport including DAW, KP, dry/wet deposition, and long-range transport potential [12, 20–22]. To cover all PFAAs, we tested the sensitivity of DAW with varying pKa (−4 to 4) and KAW (10−4 to 105) suggested (Fig. 1). At a given pKa, the DAW value increases proportionally to 1:1 as KAW value increase. The suggested KAW range (log KAW = −2.37 to 2.57 for PFOA and −2.4 to 3.29 for PFOS) produced the differences of 104 and 105 for DAW, respectively. Similarly, DAW value at a given KAW increased in proportion to the increment of pKa; that is, the pKa ranging from −4 to 4 yielded the variation of eight orders of magnitude in DAW value. This implied again that the use of the more accurate values for pKa, pH and KAW could be very important.

In the present study, we induced more reasonable values for KAW and pKa from DAW values which were calculated from PFAA concentrations measured in air vapor and rainwater. In calculating DAW (= Cair vapor /Crainwater ), it was assumed that the solely neutral species in air vapor phase and both neutral and ionic species in rainwater were present because the ionic species are nonvolatile: for example, log KAW of PFOA is −2.37 for neutral species (at suggested minimum value) versus −6.5 for ionic species [38]. The measured DAW values in this study ranged from 7.9 × 10−8 to 8.4 × 10−6 as for all investigated PFAAs (Table 3). When based on measured DAW, thus log KAW values should be within −4.0 to 0 for PFOA and −2 to 2 for PFOS as for their suggested pKa ranges. This indicated that at least the maximum values of KAW which were suggested by Kim et al. [37] would be not supported by our measurement. Two laboratory studies reported the KAW values of 0.001 [39] and 0.004–0.007 [40], which were a good agreement with COSMOtherm-modeled data within less than an order of magnitude. So, the COSMOtherm-modeled values [36] were used for KAW values of individual PFAAs in this study in order to maintain systematic consistency among the estimates of KAW. As the next step, we adjusted pKa values (at COSMOtherm-based KAW and pH = 4.5) to produce the best fitting (within ± 1%) to the measured DAW (Table 3). The estimated pKa values in this study were close to the minimum values of previously suggested values except for PFOS which was greater than the suggested maximum value but were slightly higher than the estimates of SPARC.

Estimated pKa from Observed DAW of PFAAs between Air Vapor and Rainwater
Under the typical atmospheric condition (relative humidity = 50% and TSP = 60 μg/m3), the combination of the adjusted pKa values and the minimum KAW values suggested in literature predicted the 25% and 75% of PFOA and 33% and 67% of PFOS in air vapor and water phase, respectively. On the other hand, the use of the maximum value of suggested KAWs yielded the presence of almost 100% of PFOA and PFOS in air vapor phase. Similarly, the major phase of PFAA might significantly change within the suggested pKa range of −0.21 to 3.8 for PFOA and −3.3 to −0.14 for PFOS. For instance, it was predicted under the same typical condition with their adjusted KAWs that the majority of atmospheric PFOA and PFOS might exist in water phase when their minimum pKa values were applied while 99% of PFOA and 4.5% of PFOS in atmosphere might exist in air vapor phase when their maximum pKa values were used. Again, this indicated that use of inappropriate values for pKa and KAW would cause significant errors in prediction of partitioning among three phases and related fate and transport.
3.3. Estimation of KP from Measured KP ′
The KP′ was calculated using Eq. (3) from TSP-normalized total concentrations (a sum of neutral and ionic species) measured from aerosol and the concentration measured from air vapor phase (i.e., gaseous concentration of neutral species). The log KP′ values measured in this study, ranging from −2.27 ± 0.26 for PFOS to −1.65 ± 0.47 for PFDoDA, were in very good agreement with log KP ′ values measured at a semi-urban location in Toronto, Canada [11] and at Lake Chaohu, China [13] (Table 4). It is particularly noticeable that the log KP ′ values we obtained most closely matched the values measured by annular diffusion denuder sampler used to avoid the sampling artifacts [11].

Measured Gas-particle Partition Coefficients (Log KP′ versus Log KP)
According to Eq. (4) and (8-1), the KP can be induced by multiplying KP ′and the fraction of neutral species on air particle to all species in aerosol (αneutral on particle ′). Our log KP values ranged from −8.28 ± 0.27 (for PFNA) to −6.66 ± 0.23 (for PFOS). Contrary to KP′, the KP values showed the differences by an order of magnitude between different studies (Table 4). Unfortunately, Ahrens et al. [11] used the fraction of neutral species in water phase to all species in water phase instead of αneutral on particle ′. So, the difference between measured KP values may originate from the equation error. Use of different pKa values, for which SPARC-modeled value was used in in Ahrens et al. [11], can be another possible cause of KP difference.
3.4. Dependency of Atmospheric Fate on pH and Relative Humidity
A pH of 4.5, a typical value measured at urban area, was used in this study. Acid rain monitoring showed the variation of pH of 3.2–5.4 in the study area [27, 41]. Rain pH can be changed by acid-causing gases (ACGs) like SO2, NOx, and CO2 as well as by particulate matter (PM), causing large variation as shown evidently in observed pH values (from < 4.5 to 7.5) [42].
Fig. 2 shows the pH-dependence of the occurrence of PFAAs in each of three phases. Under the meteorological condition of rH = 50% and TSP = 60 μg/m3, the pH = 4.5 yielded that > 90% of individual PFCAs (90% of PFOA) and 86% of PFOS might exist in water phase, 0.2–10% of PFCAs (10% of PFOA) and 14% of PFOS in vapor phase, and < 1% of PFCAs and PFOS in particle phase. The water phase fraction of PFAAs, mostly ionic species, can increase with increment of pH and then reach more than 99% above pH = 6.0. It was predicted that drastic change in the fraction of PFAAs could occur in the lower pH condition. At pH = ~3.5 and ~3.7, a half of PFOA and PFOS may exist in vapor phase and another half in water phase. More acidified condition (i.e., more severe atmospheric pollution by ACGs) can cause the prevalent vapor phase (> 99%) of PFOA at pH = 1.3 and of PFOS at pH = 1.5. Consequently, PFAAs in ACG-polluted air of urban/industrial area, i.e., at low pH air condition, can be moved via gaseous diffusion and wind-driven gaseous transport to other region and then can reside more shortly in the environmental system of the emitted region. On the other hand, PFAAs in rural area, of which air may be less polluted by ACGs and then be at relatively high pH (mostly, > ~5.4), can reside much longer in the environment of the domain system because the majority of individual PFAAs may exist in water phase (e.g., clouds and fogs) and can fall down via deposition of atmospheric water (e.g., rainfall and snow). Moreover it is difficult for deposited PFAAs to be evaporated considering the higher pH condition of freshwater (pH = ~7.5) where non-volatile ionic species may exist exclusively. In short, PFAAs can distribute ubiquitously and relatively uniformly from urban to rural regions by their pH-dependent atmospheric fate.

Fraction of neutral and ionic species of PFOA (a) and PFOS (b) adsorbed on air particle, in air vapor, and in water phase with pH values. A vertical solid line indicates pH = 4.5.
The volume of atmospheric water is extremely variable in time and space with the meteorological condition. Dependency of phase fractions of each species on relative humidity was presented in Fig. 3. Increase of relative humidity is inevitably associated with increasing occurrence of PFAA in water phase. In > 40% of rH, the most of PFOA and PFOS (i.e., > 80%) may exist in atmospheric water phase. However, the more than 1% of PFAAs (e.g., 5% of PFOA and 7% of PFOS) can be still present as a neutral species in vapor phase even in water-saturated condition such as fog. Vapor phase fraction of PFOA and PFOS may be more abundant below 5% and 8% of rH, respectively. More fluorinated PFCAs (> C10 PFCAs) were predicted to exist mostly in a vapor phase (> 50%) under the perfectly dried condition (< 2% of rH). Air particle-phase of PFAA accounted for < 0.1% in any cases with rH variation and exceeded 1% only for an extreme condition such as TSP = 5,000 μg/m3 and rH = 0%. This indicates that dry deposition and rain scavenging of particle-adsorbed PFAAs are likely negligible.
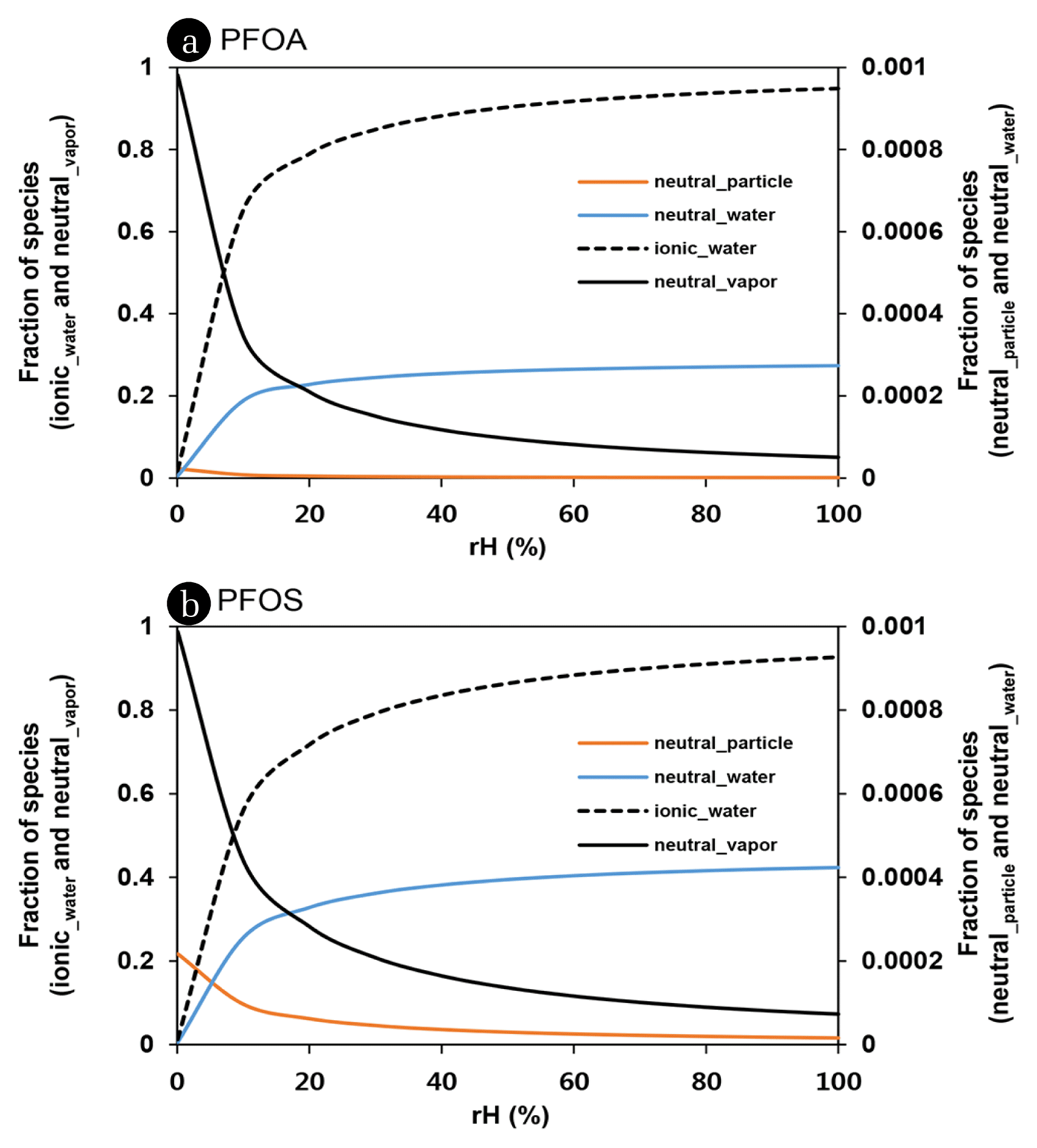
Fraction of neutral and ionic species of PFOA (a) and PFOS (b) adsorbed on air particle, in air vapor, and in water phase with relative humidity.
4. Conclusions
Environmental fate of ionizable organic pollutants such PFAAs strongly depends on their water-phase associated physicochemical properties such as pKa, pH, and KAW. So far a wide range of values for the parameters (~10,000 times difference between minimum and maximum values of pKa and KAW) have been suggested, could be significant factors enlarging the uncertainty in the prediction of atmosphere-associated fate and transport including DAW, KP, dry/wet deposition, and long-range transport potential. The result of our sensitivity analysis showed that the major phase of PFAAs present in atmosphere could significantly change within the range of suggested values for the parameters, indicating that the accurate values for the parameters are essential in assessing atmospheric and sequential fate. So, we induced the pKa and KAW values by adjusting to in situ measurements of DAW between air vapor phase and rainwater, and estimated KP of PFAAs using in situ measured gas-aerosol partition coefficient and obtained parameter values.
Our study is expected to have some implications in prediction of the environmental redistribution of other ionizable organic compounds since the atmospheric three-phase partitioning of ionizable organic pollutant is strongly associated with their redistribution among environmental compartments via volatilization, dry/wet deposition, rainfall scavenging, advection, and sorption to terrestrial surface. The redistribution of ionizable organic pollutants through atmospheric fate can be of particular importance in the coastal region where those pollutant can move backward, i.e., from the sea to the land. Our study will be helpful to understand such a fate of ionizable organic pollutants.
Acknowledgments
This work was supported by Incheon National University (International Cooperative) Research Grant in 2014.