1. Introduction
Due to the high toxicity, persistence and carcinogenicity of heavy metals, global concerns of contaminated soil by heavy metals are growing [1]. Among them, cadmium (Cd), arsenic (As), lead (Pb) and chromium (Cr) are the most familiar pollutants in heavy metal contaminated soil [2]. Various studies have shown that human activities are one of the main reasons for the formation of heavy metals in the environment [3, 4]. Extensive industrial and agricultural events such as the manufacture of pyrotechnics and electronic products, as well as the usage of Cr-containing phosphate fertilizers, can cause the severe excessiveness of Cr [5, 6]. Accumulation of As in soils is due to the usage of As-containing pesticides, the combustion of fossil fuels, the dumping of tailings and industrial wastes [5–8]. Unlike other contaminants, microorganisms or chemicals cannot degrade them once the heavy metals enter the soil environment [9, 10]. Heavy metals mainly affects the liver, kidneys and lungs of human and may also cause dermatitis and skin ulcers [11]. With the increasing public demand for environmental health, the remediation of heavy metal contaminated soil has been put on the agenda, and more and more attention has been paid by the academic community.
Researchers have researched and developed a variety of techniques for the remediation of heavy metals in contaminated soil, especially flushing [12], phytoremediation [12, 13], excavation and stabilization [5]. Traditional ex-situ restoration methods such as soil replacement and flushing are often cost prohibitive, and require significant capital investment in labor and material resources. Various reports have indicated that electrokinetic (EK) remediation is one of the most potential in-situ technologies for the treatment of heavy metals contaminated soil, such as Cu, Zn, Pb, Cd, Cr, Ni, Hg, As [14–16]. Zhou et al. [17] used electrodynamic method to remediate Cu/Zn co-contaminate soil, and the removal efficiency of Cu and Zn reached 63% and 65% respectively. However, EK can only make the pollutants in the soil move to the designated region, but cannot eliminate the pollutants. Therefore, EK is often combined with other technologies to achieve the effect of migrating and removing pollutants in the soil. For example, Afshinnasiri et al. [17] used EK in combination with PRB and EDTA to remove 70% of Cr in the soil. Ali Baratifardin et al. [18] combined the use of EK with anionic and non-ionic surfactants, which can improve the kerosene removal efficiency by 37% compared with the traditional use of EK. Sajadadhami et al. [19] used electrokinetic method combined with Fenton technology to remove 93% of phenanthrene in the soil, and the removal efficiency was 100% higher than that of traditional electrokinetic method. Among the above combined technologies, the technology combining EK and PRB can improve the removal efficiency of pollutants and the performance of EK by accelerating the transport of pollutants, thus reducing manpower and material inputs [20–23].
A variety of materials have been developed for use as PRB reaction media, such as Fe3O4 [22], zero-valent iron (ZVI) [24, 25], red mud [26], microorganism [27], humin [28] and activated carbon [29]. However, ZVI is easily affected by pH, ion composition and other factors, which will lead to PRB precipitation in the degradation process, thus reducing its permeability, reactivity and durability [19, 28]. Layered double hydroxides (LDHs) is an increasing concern due to their low cost and long-term operability as PRB materials [21, 30]. Meanwhile, recent studies have found that LDHs also has the ability to remove oxyanion contaminants. Among them, calcium-based LDH has received widespread attention due to its low cost and easy availability. Li et al. [31] found that CaAl-LDH has an excellent adsorption capacity of Cr (VI) and U (VI), and the maximum adsorption was up to 104.82 ± 0.02 mg/g and 54.79 ± 0.02 mg/g, respectively. Lu et al. [32] used CaFe-Cl-LDH and CaFe-NO3-LDH to adsorb arsenate. The maximum adsorption capacity was 150.5 mg/g and 148.0 mg/g, respectively. Common calcium-based LDHs are CaFe-LDH and CaAl-LDH. However, no studies have compared the application of CaFe-LDH and CaAl-LDH to the remediation of heavy metal contaminated soils. Therefore, this article aims to use low-cost and easy-to-obtain CaAl-LDH and CaFe-LDH combined with EK to remediate heavy metal contaminated soil.
Numerous studies have evaluated the potential of EK-PRB techniques to treat heavy metals contamination in soil. However, most of the surveys were carried out on soils contaminated with single metals. The previous work of our group [14, 21] only explored the remediation of single metal (Cr) contaminated soil by EK-PRB technology in detail. Cappai et al. [26] enhanced the removal of Cr and As by transforming red mud (TRM) into an active material for PRB, and evaluated the synergic interaction between the EK and the PRB system. Yuan et al. [33] used cobalt-supported carbon nanotubes (CNT-Co) as PRB active materials to study EDTA-enhanced EK-PRB remediated As (V) contaminated soil, and the removal efficiency was 63% after 5 d of remediation. The removal efficiency of As was only 35%. Currently, using LDHs to remediate polymetallic contaminated soil is relatively rare. From the foregoing, oxygen anions can be adsorbed by LDH. The common heavy metals in the soil in the form of oxygen anions are As and Cr. Therefore, this article explores the remediation of As/Cr co-contaminated soil by CaAl-LDH and CaFe-LDH.
In summary, the purpose of this study is to investigate (i) the effect of PRB material, PRB position, initial voltage gradient and run time on the removal efficiency of As and Cr in the EK-PRB synergistic system, (ii) the migration direction and behavior of As and Cr in EK-PRB systems.
2. Materials and Methods
2.1. Materials
The commercial kaolin soil was used in this research. The contaminated soil with As (III) (100 mg/kg) and Cr (VI) (500 mg/kg) was prepared by sodium arsenite and potassium dichromate. In detail, commercial kaolin was placed into beaker with distilled water (the ratio of soil to water is 2:1), then a mixture of sodium arsenite and potassium dichromate was added. The soil sample was placed in a dry and ventilated place for 30 d. The PRB media were CaAl-LDH and CaFe-LDH, which were prepared by co-precipitation method [34]. The prepared LDH material was repeatedly washed 2 – 3 times with nitrogen-filled distilled water, dried at 105°C, then passed through a 100-mesh sieve and stored for use.
2.2. Experimental Setup and Procedures
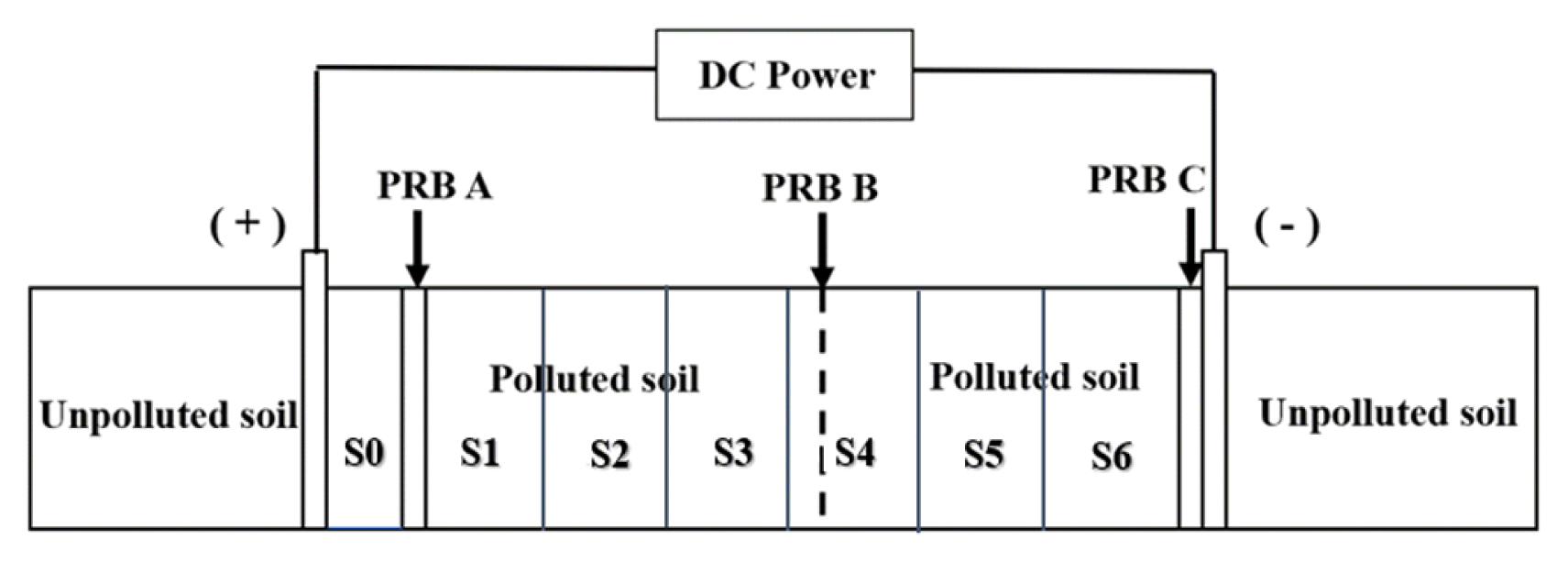
The EK-PRB experiment was performed in a laboratory scale plexiglass apparatus consisting of plexiglass, electrodes, direct current (DC) power Supply, soil and PRB. The schematic diagram of the EK-PRB device (250 mm × 100 mm × 100 mm in length × width × height) used in this experiment is illustrated in Fig. 1. A total of 7 sampling regions (S0–S6) were divided from anode to cathode. Two flaky graphite electrodes were vertically inserted into the uncontaminated soil, and when energized, they serve as the anode and the cathode, respectively. In every operation, Cr-As co-contaminated soil was filled (about 4 cm). However, the As and Cr-free soil was placed into S0 region, anode and cathode. A, B and C were the filling sites for active material of the permeable reactive wall in the experiment. 8.1 g of active material was filled corresponding to the contaminated soil concentration, and the LDH was fixed in the soil using nylon cloth and acrylic porous plate. The thickness of PRB was maintained at 6mm.Adjust the voltage of the DC power supply to control the soil to constant voltage. The way that the DC power supply was regulated and the voltage applied to the soil were consistent with our previous work [14].
In this experiment, the plate-shaped graphite electrodes were directly inserted into the wet soil. Soil moisture contents of all tests were 30% according previous report [21]. The removal efficiency of TCr and TAs by different PRB insertion sites (A, A + B, A + C), voltage gradients (1, 1.5 and 2 V/cm), running time (24, 48, 72, 96 and 120 h) and different PRB materials (CaAl-LDH and CaFe-LDH) were investigated. All experiments are shown in Table 1.
2.3. Analytical Methods
During the entire experiment, the soil conductivity was measured by a multimeter (Model VC890C+, Shenzhen Victor Hi-Tech, China). The calculation of current density refers to the previous work [14]. The soil pH was determined by a pH meter (Model LI-120) and a glass electrode (Model CL 51), with the soil to water ratio of 2:1. To determine TAs and TCr, 0.15 g of dry soil was digested in 10 mL of HNO3 and 2 mL of HClO4, then digested in 5 mL of HF and 2 mL of HClO4 until the solution was clear and transparent. The specific concentrations of obtained TCr/TAs were measured by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) (Model Prodidy, Leeman). The XRD patterns were obtained using a Dmax/RB diffractometer (Rigaku Co.) in which Cu Kα radiation (λ = 0.15406 nm) was carried out at 40 kV and 100 mA. Approximately 1.5 mg of the sample was uniformly mixed with 300 mg of KBr (Uvasol) and compressed under vacuum to make 1.3 cm particles[35], and then FTIR spectra were obtained by FTIR 380 (Thermo Fisher Scientific).
3. Results and Discussions
3.1. Effects of PRB Materials on EK-PRB Systems
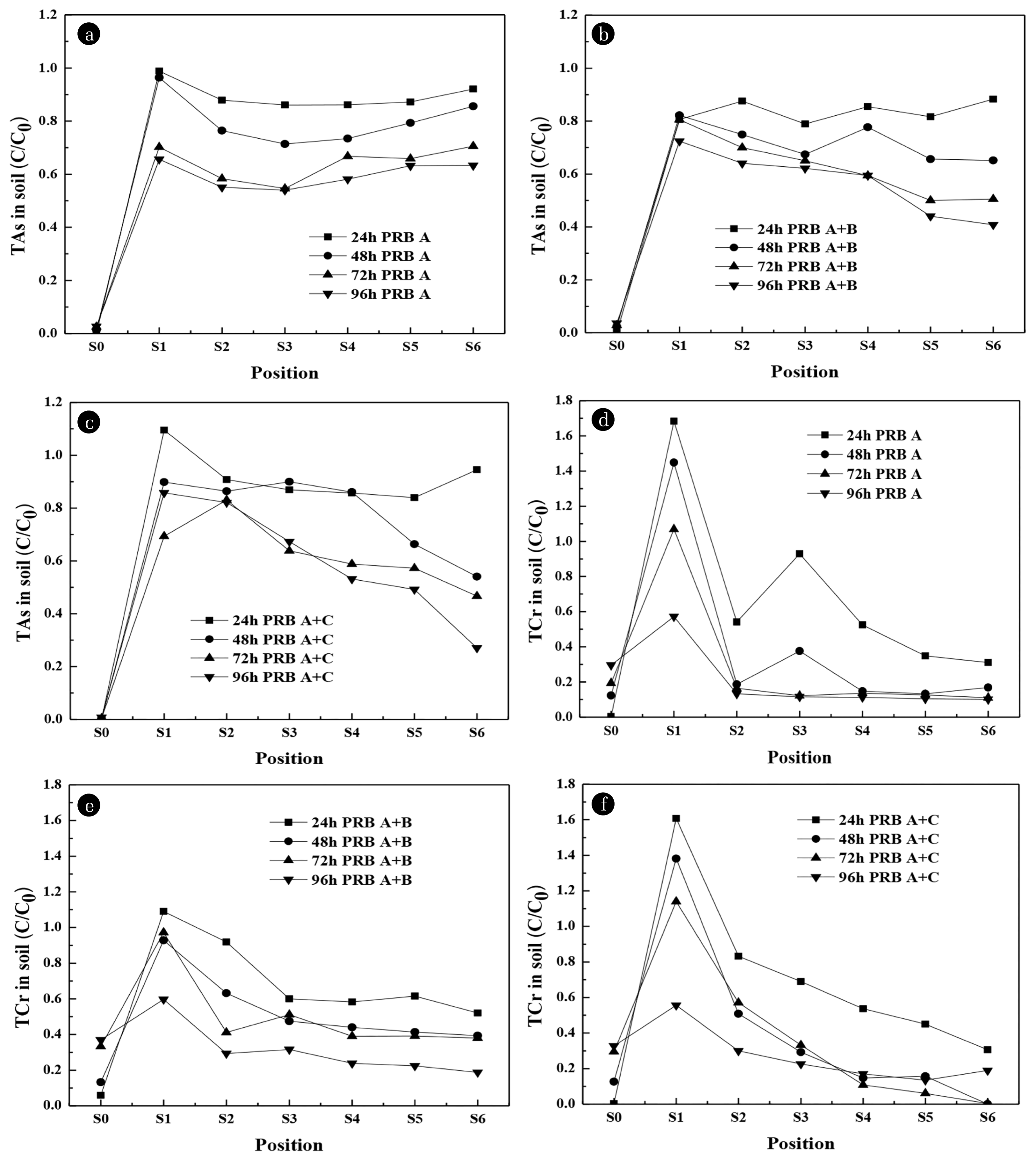
To investigate the effect of PRB materials on the concentration of TAs and TCr, TAs and TCr concentration in soil with different PRB materials after 24, 48 and 72 h are shown in Fig. 2.
The concentration of residual TAs is shown in Fig. 2(a). In CaAl-LDH system, S1 region, the accumulation of TAs was 98.8% in 24 h; In S1 – S6 regions, the concentration of residual TAs decreased with the increase of time, indicating that As migrated to the location of PRB and was adsorbed by PRB. In contrast, in the CaFe-LDH system, the accumulation efficiency of TAs in the S1 region within 24 h was 88.9%. In the S1 – S6 regions, except for the S1 region (48 h), the residual amount of TAs decreased with the increasing of time, which was caused by the dissolution of CaFe-LDH in S1 region (48 h). In Fig. 2(c) and 2(d), the increase of calcium content and pH value in S1 region (48 h) also confirmed the dissolution of LDH. Except for S1 region (48 h), the concentration of residual TAs in CaAl-LDH system was higher than that in CaFe-LDH system in 24 h and 48 h.
For 72 h, except for S6 region, the TAs concentration in other regions of CaAl-LDH system was lower than that of CaFe-LDH system. It was because As in the S6 region of the CaAl-LDH system could not migrate to the PRB location in time and was captured by the PRB. Within 72 h, the removal efficiency of TAs in CaAl-LDH and CaFe-LDH systems were 35.6% and 34.2%, respectively. During 48 h to 72 h, the removal and migration of As in CaFe-LDH system were significantly lower than that in CaAl-LDH system. The reason was that CaFe-LDH dissolved obviously [35], leading to the release of As, which could not be captured by PRB in time. In Fig. 2(c) and 2(d), the increase of calcium content and pH value of CaFe-LDH at 72 h also confirmed the dissolution of LDH [35].
The concentration of residual TCr is shown in Fig. 2(b). In the S1 region of the CaAl-LDH system, the accumulation efficiency of TCr within 24 h was 168.3%. This indicated that Cr near the cathode moved toward the anode and accumulated in the S1 region. Then the concentration of residual TCr decreased with the increasing of time in S1 – S6 regions; after 72 h, the remaining TCr in the S3 – S6 regions accounted for 12%. By comparison, in CaFe-LDH system, the accumulation of TCr was 84.3% in 24 h; then the concentration of residual TCr decreased with the increasing of time; however, about 30% Cr was remnant in S3 – S6 regions. The removal efficiency of TCr was 71.2% and 65.0% in 72 h, respectively. It could be found that parts of Cr accumulated in soil and did not move to the anode in time.
Two experimental groups were designed to explore the effects of different Ca-based LDHs during the remediation process by EK-PRB. The relationship of Ca concentrations and remediation time of all regions are shown in Fig. 2(c).
The Ca concentration in each region indicated the dissolution of LDH and the migration direction of Ca. In both Ca based LDH systems, Ca concentration reached the highest near the anode, and gradually decreased from S1 to S6 regions. In CaAl-LDH experiments, there was a slight increase of Ca concentrations with the increasing of time extension in each region. Compared with CaAl-LDH, the Ca concentration in CaFe-LDH experiment was significantly increased and finally migrated to the cathode. Based on above findings, it could be concluded that the migration of Ca to the cathode would happen. Moreover, it was easier for Ca to dissolve out in CaFe-LDH experiments than in CaAl-LDH.
The changes of pH of every region after running for different time with different PRB is shown in Fig. 2(d). The soil pH of CaFe-LDH system was much higher than that of CaAl-LDH system. The pH of S1 region was 6.43 (24 h), 6.82 (48 h) and 6.22 (72 h). The reason was that there was a large amount of CaFe-LDH dissolution in the CaFe-LDH system [36], which caused the increase of soil pH. In the two systems, the soil pH value in S0 region decreased with the increasing of time, while the soil pH value in S6 region increased.
3.2. Effect of the PRB Position on EK-PRB Systems
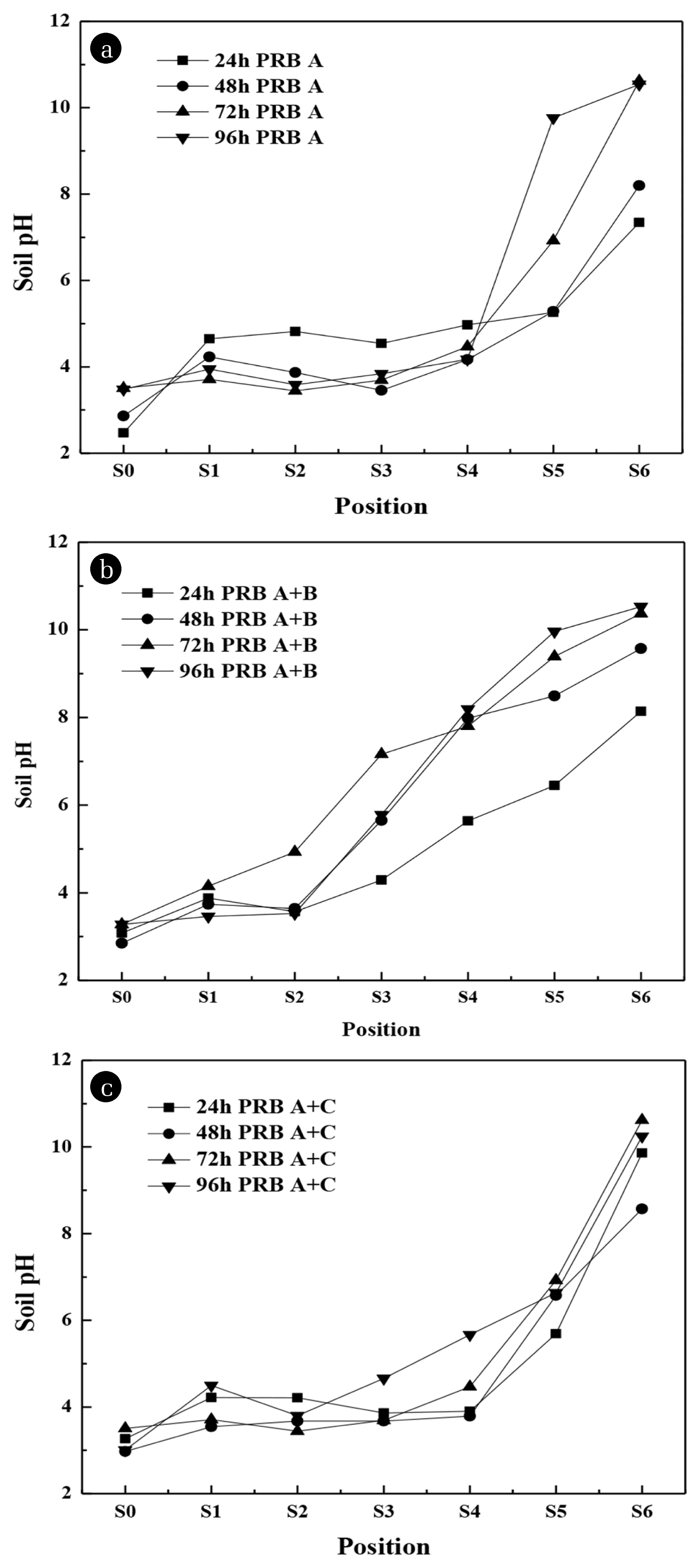
Fig. 3 and Fig. 4 show the concentration distribution of the two heavy metals and the change of soil pH respectively when PRB was placed in different locations.
In PRB A system (Fig. 3(a)), with the extension of operation time, the concentration of residual TAs in soil (S1 – S6 regions) decreased gradually, and the middle (S2 – S4 regions) decreased rapidly. Compared with PRB A system, the concentration of residual TAs in PRB A + B system gradually decreased (Fig. 3(b)), while the concentration of residual TAs in S4 – S6 regions was less than 40% – 65% within 96 h, because LDH placed in B position (middle) could capture heavy metal in time. In the PRB A + C system (Fig. 3(c)), the concentration of residual TAs fluctuated in most sample regions, because PRB C system could fix a part of As. The residual TAs in S5 – S6 regions were less than 20% – 45% within 96 h. After 24, 48,72 and 96 h treatment, the removal efficiency of TAs were 10.3%, 19.6%, 35.6% and 40.1% in PRB A system, 16.3%, 27.8%, 37.4% and 42.9% in PRB A + B system, and 8.1%, 21.2%, 36.8% and 39.2% in PRB A + C system. It can be seen from Fig. 4 that the final pH of contaminated soil in the S1 – S4 regions was 3.5 – 6, and the pH of S5 – S6 regions near the cathode was above 7.
From the Eh-pH diagram of As, when pH is 3.5 – 6, As (III) mainly exists in the form of H3AsO3, part of As (III) in the soil will be transformed into As (V), and As (V) mainly exists in the form of H2AsO4−, when pH is above than 7, As (V) mainly exists in the form of HAsO42−, while PRB mainly adsorbs As in the form of anions, which can explain the effect of different PRB placement positions on As removal in different soil regions. Within 0 – 48 h, the removal efficiency of TAs in the PRB A + B test was relatively high, which indicated that the remediation of PRB in B position was timely. With the increasing of time, the removal efficiency of the three groups were basically the same, which indicated that the PRB location had little effect on the removal of TAs in soil. It meant that multiple PRBs did not show higher utilization with the same quality LDH material.
The distribution of TCr concentration in the soil with different PRB positions after 24, 48, 72 and 96 h are shown in Fig. 3(d), 3(e) and 3(f). In PRB A system (Fig. 3(d)), the concentration of residual TCr in S2 – S6 regions decreased rapidly with the increasing of time, the concentration of residual TCr was 92.8% (24 h), 37.6% (48 h) and 12% (72 – 96 h) in S3 region which was caused by the local accumulation of Cr in this region from S4 – S6 regions. Compared with PRB A system, the residual TCr concentration in the PRB A + B system decreaseed slowly with the increasing of running time (Fig. 3(e)); because the LDH placed in the B position (middle) can capture pollutants in time, the concentration of residual TCr was 109.0% (24 h), 92.8% (48 h), 97.1% (72 h) and 55.6% (96 h) in S1 region. In PRB A + C system (Fig. 3(f)), the concentration of residual TCr decreased at the slowest rate and was much lower in S5 – S6 regions; Due to the setting of multiple PRBs to form a certain resistance, the migration of Cr was blocked; the concentration of residual TCr of S1 region was 160.8% (24 h), 141.1% (48 h), 114.0% (72 h) and 59.6% (96 h). After 24, 48, 72 and 96 h treatment, the removal efficiency of TCr was 27.7%, 59.0%, 71.2% and 81.0% in PRB A system; 27.9%, 45.3%, 49.1% and 69.1% in PRB A+B system; 26.3%, 58.6%, 63.1% and 73.7% in PRB A + C system. The removal efficiency of TCr is similar in the first 24 h, but with the increasing of running time (48 – 96 h), the removal efficiency of PRB A system was the best. It was demonstrated that the PRB A could reduce the concentration of TCr in part of regions, while the PRB B hinder the migration of Cr.
The soil used in the experiment was kaolin soil mixed with hexavalent chromium (Cr (VI)) and trivalent arsenic (As(III)). According to the migration and removal of As and Cr in the compound contaminated soil, it could be found that the migration of As tends to move to the two poles, and the migration of Cr tended to move to the anode. The migration of As and Cr in soil mainly occurred in soil solution under electrokinetic action. The final pH of contaminated soil in the test system was 3.5 – 6, and the pH of S5 – S6 regions near the cathode was above 7.
From the Eh-pH diagram of As, in our system, As (III) mainly exists in the form of H3AsO3, part of As (III) in the soil will be transformed into As (V). As (V) mainly exists in the form of H2AsO4− and HAsO42−[37]. According to the Eh-pH diagram of Cr, Cr (VI) mainly exists in the form of Cr2O72−[38]. PRB was easy to capture Cr (VI), and part of Cr (VI) in soil was transformed into Cr (III). Cr (III) was formed by the interaction of soil organic matter and microorganisms [14]. Cr (III) in soil was easy to be adsorbed by soil particles or form hydroxide, which limited the movement of Cr to cathode. The anions in soil solution tend to migrate to the anode through electromigration, and the non-ionic substances tend to migrate to the cathode through electroosmosis [22]. Therefore, As migrated to the two poles, which affected the directional migration and the removal of TAs; while most Cr migrated toward anode, which was conducive to the removal of Cr.
3.3. Effects of initial Voltage gradient on EK-PRB Systems
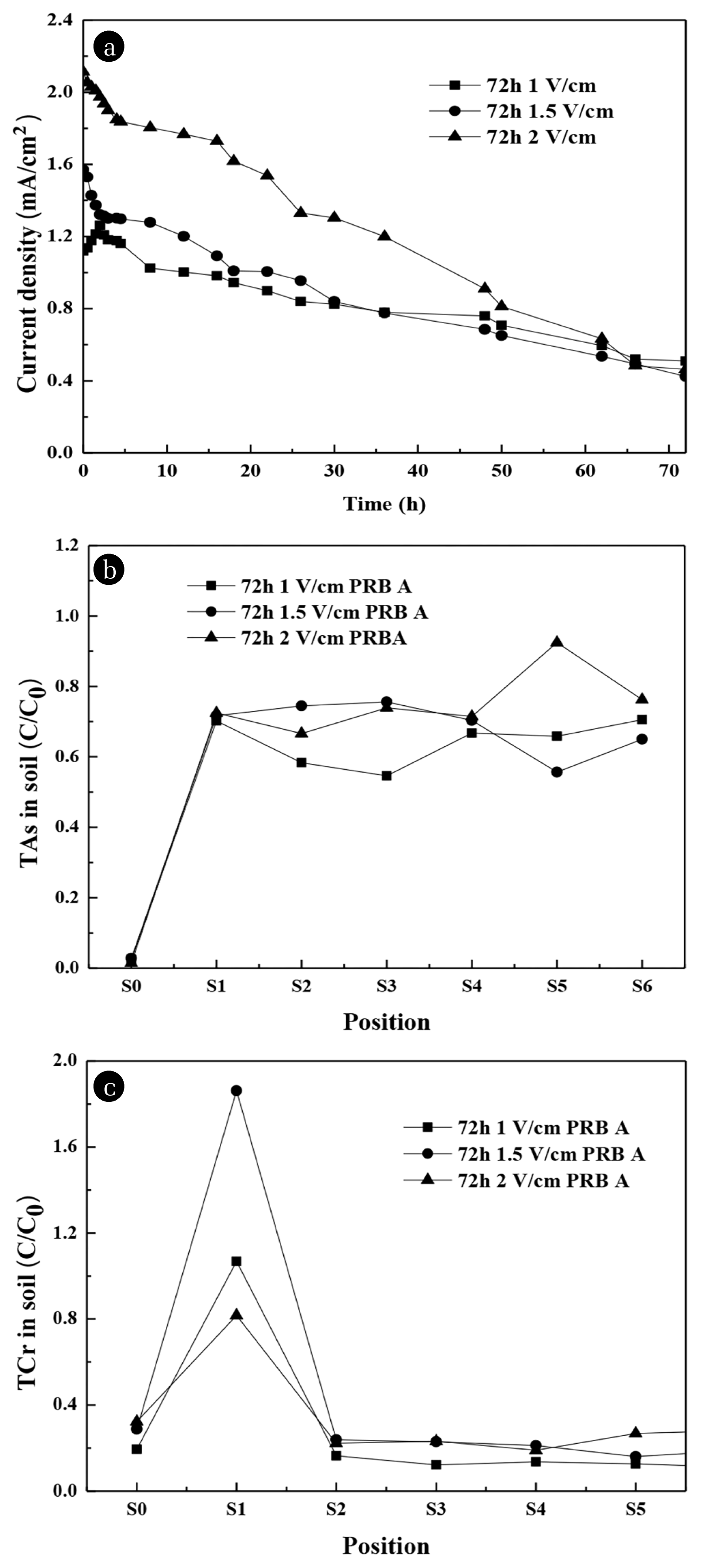
The variation of current density of the device during 72 h operation under different voltage gradients are shown in Fig. 5(a). In general, the current density increased as the voltage increased. When the soil constant voltage was 1, 1.5, 2 V/cm, the initial current density of the soil was 1.12, 1.57 and 2.11 mV/cm2, respectively. In the first 2 h, when the constant voltage was 1 V/cm, the current density of the soil increased to 1.26 mV/cm2; when the constant voltage was 1.5 and 2 V/cm, the current density of the soil decreased to 1.32 and 1.97 mV/cm2, respectively. At 72 h, when the constant voltage was 1 V/cm, the current density of the soil was reduced to 0.51 mV/cm2; when the constant voltage was 1.5 and 2 V/cm, the current density of the soil continued to decrease to 0.42 and 0.46 mV/cm2, respectively. The current density tended to decrease under the high voltage gradient. Under a low voltage gradient, the current density tended to rise and then fell. When the voltage gradient was small, it took some time for the ions in the soil to dissolve. During this time, as the dissolved electrons increased, the current density continued to rise. The lower the applied voltage gradient, the more obvious the current density would increase firstly and then decrease. This phenomenon was similar with other researchers [24, 39].
Under the conditions of different voltage gradients, the concentration of residual TAs/TCr at each soil region after 72 h are shown in Fig. 5(b) and 5(c), respectively. The concentration of residual TAs increases as the voltage gradient increased. Under the voltage gradients of 1 V/cm, the concentration of residual TAs in the S2 and S3 regions was lower (Fig. 5(b)), which were 58.3% and 54.6%, respectively; S4 – S6 regions distributed between 65% – 70%. Compared with 1.0 V/cm, S2 – S3 regions were much higher under 1.5 and 2 V/cm. Under 1.5 V/cm, the concentration of residual TAs in the S2 and S3 regions were 74.5% and 75.7%, respectively; S4 – S6 regions distributed between 55% – 70%; Among them, in the S5 region, the concentration of residual TAs was the lowest, which was 55.6%. Under 2 V/cm, As accumulated in S5 region, which reached 92.5%; the concentration of residual TAs in the S2 – S3 regions were 66.7% and 74.0%, respectively; S4 – S6 regions distributed between 71% – 92.5%. Through calculating, the average removal efficiency of TAs in the S1 – S6 regions were 35.6%, 31.2% and 24.5% under 1, 1.5 and 2 V/cm. The concentration of residual TAs was essentially zero in the S0 region under all conditions, indicating that PRB can effectively capture As.
As the voltage gradient increased, the concentration of residual TCr increased first and then decreased. It can be seen from Fig. 5(c) that the concentration of residual TCr in the S2 – S6 regions was less than 30% under the voltage gradients of 1, 1.5 and 2V/cm, and the lowest at 1V/cm. In the S1 region, the residual TCr was 106.9%, 186.2% and 81.7%, respectively. Through calculating, removal efficiency of TCr in the S1 – S6 regions were 72.1%, 51.8% and 66.5% under 1, 1.5 and 2 V/cm. Meanwhile, under different voltage gradients, the TAs content in the S0 region was 19.4%, 28.7% and 32.3%, respectively. The results showed that the Cr was more likely to penetrate PRB under higher voltage gradients, which was due to the dissolution of the CaAl-LDH material [30, 36].
3.4. Effects of Running Time on EK-PRB Systems
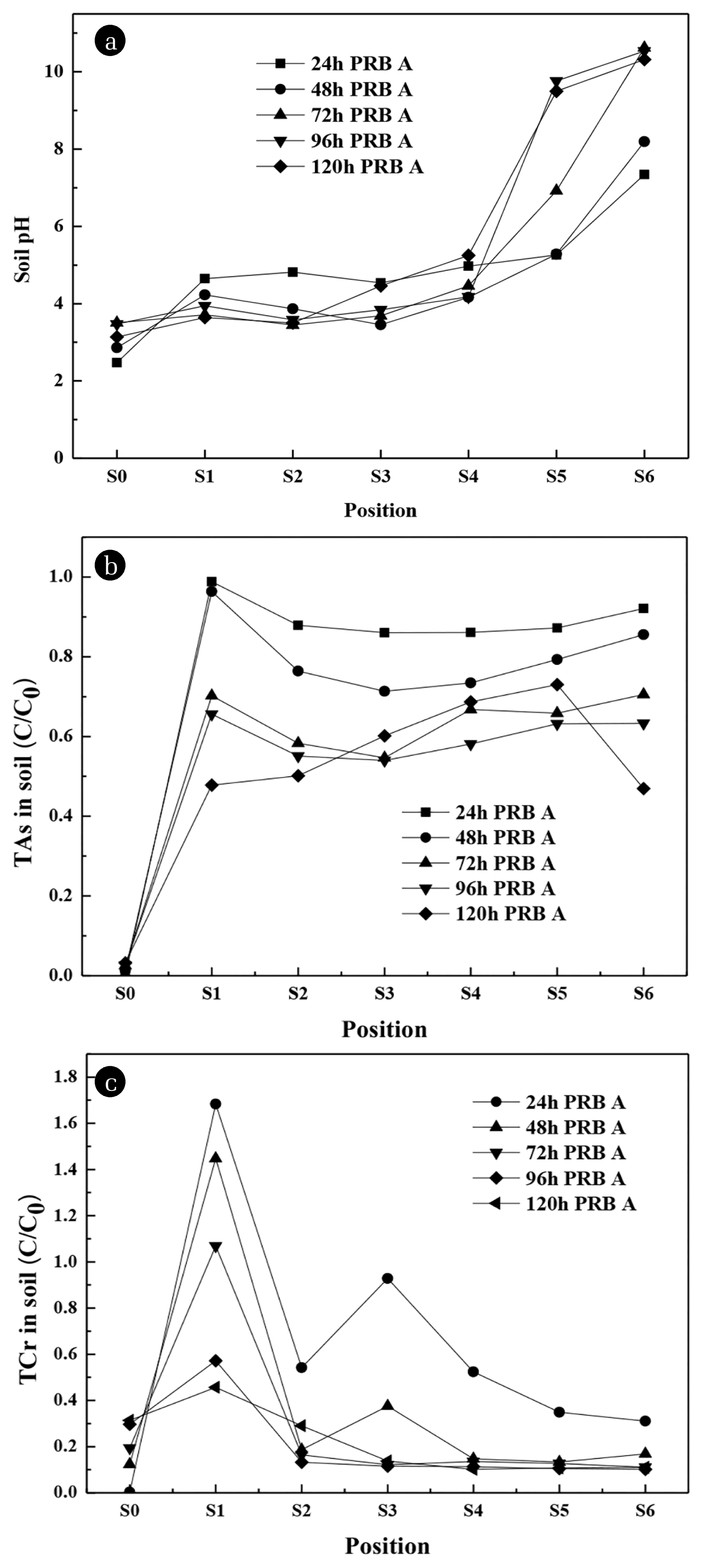
The pH changes of each soil region under different running time are shown in Fig. 6(a). At the beginning of the experiment, the pH of As/Cr-free soil and As/Cr-contaminated kaolin soil was 3.22 and 3.52, respectively. The pH of S0 region was 2.47 (24 h), then increased to 3.50 (72 h), and decreased to 3.14 at 120 h, which was caused by partial dissolution of LDH. The trend of pH was similar with the increase of time in S1, S2, S3 and S4 regions, the pH was 4.65, 4.82, 4.54 and 4.97 at 24 h, then remained between 3.5 – 5.3. The pH of S5 and S6 regions increased to 6.92 and 10.62 at 72 h; S5 region kept on rising to 9.76 at 96 h and then decreased to 9.5 at 120 h; S6 region decreased slightly to 10.32 at 120 h. Which was caused by the dissolution of the CaAl-LDH [36]. Meanwhile, during the remediation process, H+ and OHwere produced due to the presence of electric field. The pH of the soil changed due to the electrolysis of the cathode and anode. The distribution and change of soil pH are affected by the migration of H+, OH− and the initial concentration of other ions [15].
Fig. 6 shows the changes of residual TAs/TCr concentration with the increasing of time in every region. In Fig. 6(b), the changes of TAs were similar for S1 – S6 regions, except S3, S4 and S5 region increased slightly. In S1, S2, and S6 regions, the concentration of residual TAs gradually decreased with the increase of running time, which was 47.8%, 50.2% and 46.9% at 120 h, respectively. The concentration of residual TAs in the S3 – S4 regions decreased to 54.0%, 58.1% and 63.2% at 96 h, and increased to 60.2%, 68.7% and 73.0% at 120 h, respectively. The concentration of residual TAs in S0 region raised gradually with the increasing of time. The TAs residual amount of was relatively homogenous in every region, which further testified that the migration of As in soil did not migrate in a certain direction.
In the Fig. 6(c), the concentration of residual TCr in S0 region was 0.3% (24 h), 12.3% (48 h), 19.4% (72 h), 29.6% (96 h) and 31.3% (120 h); Due to most of Cr migrated to PRB region, it rose to 168.3% (24 h) and then gradually decreased to 45.6% (120 h) in the S1 region; In S2 – S6 regions, the concentration of residual TCr decreased with the increasing of time and was 29.0%, 13.7%, 10.2%, 10.8% and 11.0% in 120 h, respectively. The concentration of residual TCr in the S0 region gradually increased with the increasing of time and was caused by the dissolution of LDH. From the change of TCr concentration in each region with the increasing of time, it intimated that Cr tends to migrate toward the anode, mainly incorporation of Cr(VI) in the form of an anion, which tends to migrate toward the anode.
By calculation, the average removal efficiency of S1–S6 regions increased gradually during the period of 24 – 96 h, the removal efficiency of TAs was 10.3%, 19.6%, 35.6% and 40.1%, respectively and the removal efficiency of TCr was 27.7%, 59.0%, 71.2% and 81.0%, respectively. After prolonging the running time to 120 h, there was no significant increase in TAs and TCr removal efficiency, which were 42.2% and 80.0%, respectively. More As and Cr accumulated in the S0 region with the increasing of time. This could be caused by the dissolution of the CaAl-LDH and released again[40]. In previous study, it has demonstrated that CrO42− and Cl− during the dissolution-reprecipitation process in CaAl-LDH could exchange anion [30].
3.5. The Function of CaAl-LDH as PRB Materials
CaAl-LDH as PRB material could immobilize As and Cr. However, high moisture content and low pH near the anode could lead to the dissolution of CaAl-LDH, it had effects on the removal of As and Cr. The recovery of CaAl-LDH and the concentrations of As and Cr were determined at 24 h, 48 h, 72 h, 96 h and 120 h. The initial PRB content was 8.1 g. As is shown in Table S1, the recovery of PRB decreased gradually with the increasing of time. The possibility was that the CaAl-LDH dissolved in EK-PRB system and there was some loss of LDH during the recovery process[30]. In the PRB system, the concentration of TAs was 1.96 mg/kg at 24 h, then decreased to 13.17 mg/kg at 120 h; while the concentration of TCr was 393.4 mg/kg at 24 h, then increased to 1,285.3 mg/kg at 72 h, was 982.6 mg/kg at 120 h.
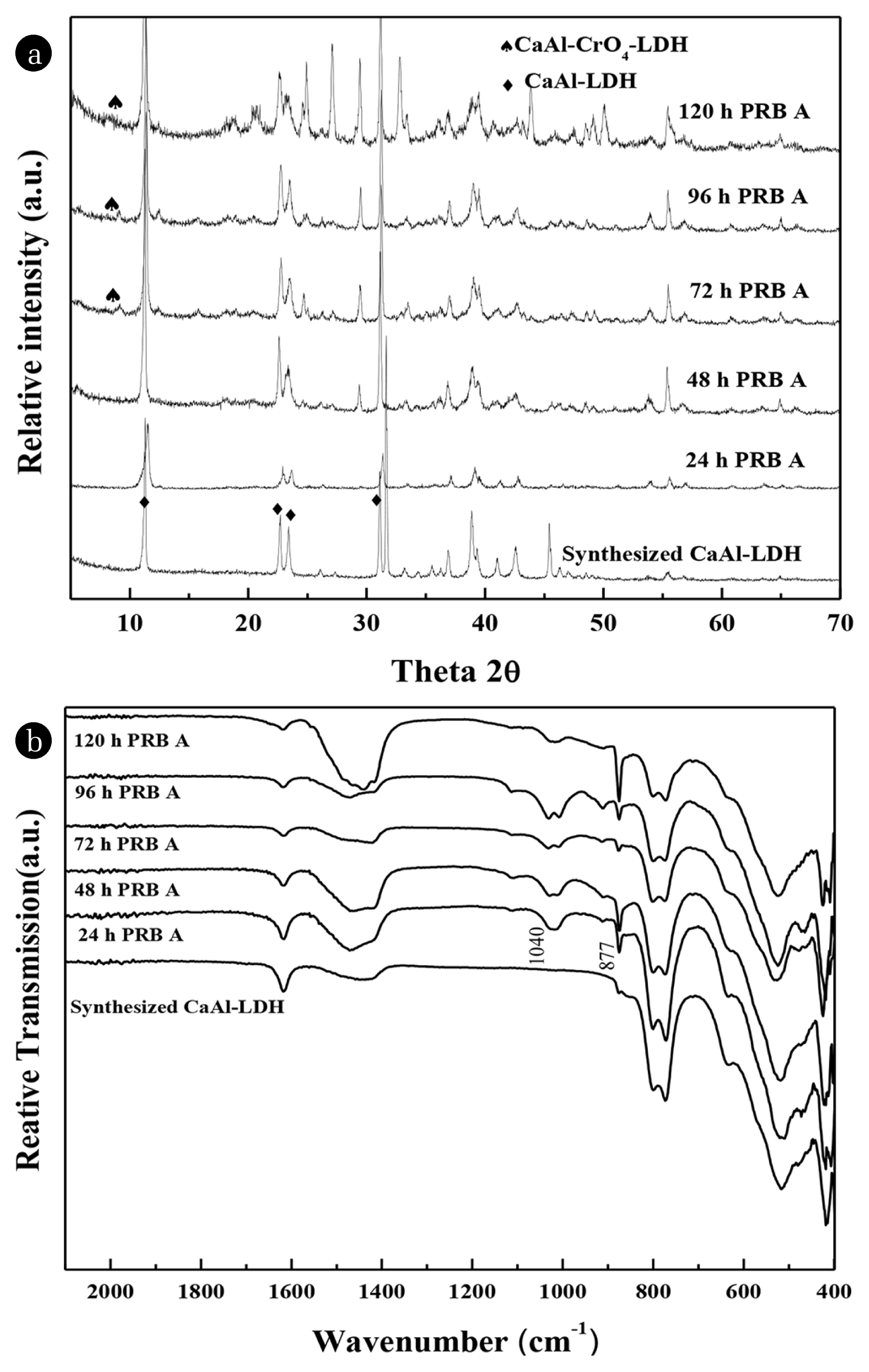
The XRD (a) pattern and FTIR (b) spectrum of CaAl-LDH before and after EK-PRB treatment are shown in Fig. 7 at different running time. According to Fig. 7(a), it was observed that the main characteristic peak (XRD) of CaAl-LDH was always maintained within 24 to 96 h. After 72 h and 96 h of treatment, CaAl-LDH (JCPDS 43-0088) crystals still retain the original crystal form, while the weak characteristic peaks (2θ = 10.520 and 21.131) of chromate hydrocalumite (CaAl-CrO4-LDH, JCPDS 42-0063) were always displayed in the PRB medium. After 120 h of treatment, the characteristic intensity of CaAl-CrO4-LDH was stronger, and more stray peaks appeared at the same time. The characteristic peaks (♠) of the new phase fitted well with those of chromate hydrocalumite (Ca4Al2(OH)12CrO4(H2O)4, JCPDS 42-0063) [14]. It was not observed significantly the insertion of As in LDH.
Fig. 7(b) compares the FTIR of LDH after different running time. The original CaAl-LDH exhibited a typical FTIR pattern described in a previous report [14], in which the broad band centered at 3,485 cm−1 was attributed to the stretching vibrations of hydroxyl groups and CaOH [41]. The peak at 1,621 cm−1 came from the deformation vibrations of water molecules [42]. The peaks at 787 and 532 cm−1 were attributed to v(Al-O) and v(Al-OH) inside CaAl-LDH, respectively [43]. Moreover, an overlapped band at 1,460 cm−1 was attributed to the v(CO32−). In addition, as the operating time of the device was extended, the vibration intensity at 3,485 cm−1 was gradually reduced due to the reaction of O-H in CaAl-LDH with As and Cr in the device. The peaks around 787 and 532 cm−1 also began to decay (from 24 to 96 h), whereas peak occurred at 877 cm−1 due to the vibration of Cr-O [44]. The width of the overlapped band at 1,460 cm−1 decreased with the increasing of running time, which could be explained by the CO32− gradual dissolution from the CaAl-LDH surface. Besides, the band at 1,040 cm−1 was a vibration of Si-O, it might be some kaolin during the recovery of PRB. Therefore, the CaAl-LDH could captured As and Cr. The removal mechanism of As by CaAl-LDH was surface adsorption, while the Cr was surface adsorption and intercalation.
4. Conclusions
The effects of PRB active material, PRB position, voltage gradient and running time on the migration and conversion of Cr and As were studied in this paper. The poor stability of CaFe-LDH resulted in a large amount of Ca dissolved and migrated to the cathode, so CaAl-LDH was selected as the PRB medium material. The location of PRB affected the migration and accumulation of As and Cr during the remediation process. When PRB was located near the anode (PRB A), the overall remediation effect was better than that of PRB A + B and PRB A + C. Under high voltage gradient, the pollutants were easier to pass through the PRB, so 1 V/cm was the best voltage gradient. The results showed that the removal efficiency of TAs and TCr were 40.1% and 81.0%, respectively at 96 h. CaAl-LDH immobilized As mainly by surface adsorption, and Cr by surface adsorption and intercalation. In this study, EK-PRB could effectively remediate the As/Cr co-contaminated soil, because EK could make As/Cr in the form of oxyanion migrate to the location of PRB, and PRB could effectively adsorb oxyanion. Therefore, the authors suggest that EK-PRB can also be applied to other oxyanion-contaminated soils. Additionally, other calcium-based substances will be considered as PRB materials in the future.