1. Introduction
The occurrence and effects of disinfection by-products (DBPs) and trace-level organic contaminants (TrOCs) have attracted growing attention of the environmental community [1, 2]. In the case of DBP controls, current regulations tend to focus on two most prominent, on the mass basis, DBP groups of trihalomethanes (THMs) and haloacetic acids (HAAs) [3, 4]. However, ongoing research shows that actual health effects may be associated with DBPs other than HAAs and THMs [5, 6]. Such species include, among others, N-DBPs (e.g., haloacetonitriles and haloacetamides) and I-DBPs (e.g., monoiodoacetic acid (MIAA)) [7, 8]. Some TrOCs groups, notably iodine-containing contrast media (ICMs) used in medical imaging [9, 10] and frequently encountered in the environment [11, 12] have been shown to be precursors of I-DBPs formed via the degradation of ICMs by halogen-based disinfectants or reactive oxygen species (ROS) formed in advanced oxidation processes (AOPs), as well as in EC treatment that involves both oxidation and reduction [13–16].
Adverse effects associated with DBPs and TrOCs and the difficulties in preventing their generation and/or removal by conventional techniques necessitate the development of alternative treatment methods. Electrochemical (EC) methods constitute a distinct group of such methods because they employ reactions induced by electric current passing across the interface between an EC-controlled working electrode, or a system of electrodes, and the solution being treated. This process can be accompanied by direct interactions of a target compound with the electrons being released or consumed at the interface. In this case the EC treatment can be termed direct. Alternatively, an EC process may result in the formation of oxidants (e.g., ROS, halogen species) or reductants that react with the target contaminant in solution bulk but not necessarily at the electrode/solution interface [17, 18]. Such mode of EC treatment can be termed indirect.
EC control of water treatment processes offers important benefits, for instance i) ease of on-demand operations and the possibility of a complete automation of water treatment, ii) little or no need to use externally stored chemicals, iii) options to control and adjust the EC potential of the working electrode(s) to optimize the degradation of target compounds and suppress background reactions (e.g., reduction or oxidation of water), and iv) a wide range of EC reactor configurations and available electrode materials, including those with electrocatalytic properties.
These aspects of EC processes as well pertinent theoretical and practical challenges have been examined in a number of prior reviews and related publications, for instance [18–20]. Given the vast amount of data generated in the studies of EC-driven processes relevant to water treatment, this review will henceforth focus on two specific EC applications, namely on the EC-controlled reductive dehalogenation of DBPs and EC degradation of ICMs.
2. EC-controlled Direct Reductive Dehalogenation of DBPs
2.1. Reductive EC Dehalogenation of Haloacetic Acids
Reductive EC dehalogenation (that is, the EC reactions that cause the cleavage of halogen atoms from the parent molecule) of HAAs which constitute an important class of DBPs has been addressed in considerable detail over the last two decades [21]. In a relatively early study, Korshin and Jensen [22] showed that di- and trihalogenated chlorine- and bromine-containing HAAs exhibit notable EC activity at the surface of cathodically polarized gold and copper rotating disk electrodes (RDE). The current associated with the EC reduction of HAAs was considerably higher in the case of copper. The EC reduction currents increased with the number of halogen atoms in HAA molecules. All HAAs except monochloroacetic acid (MCAA) were found to be susceptible to EC dehalogenation which was especially rapid from Br-containing HAAs. The formation of MCAA with its low EC activity yet a potentially considerable toxicity was judged to be a limiting factor in the developed of EC methods to removes HAAs.
These trends were in agreement with those observed in Hozalski et al. [23] for the case of the reduction of trihalogenated HAAs by zero-valent iron (ZVI), which is widely present in drinking water distribution networks and other engineered systems. (While the reduction of HAAs or other DBPs/TrOC by ZVI is not explicitly EC-controlled, it involves the coupling of the EC rate of Fe oxidation and that of the reduction of the involved contaminants or dissolved oxygen. As such, these processes can be considered to constitute a subtype of EC reactions). Experiments with trichloro-, tribromo-, dibromochloro- and bromodichloroacetic acids (TCAA, TBAA, DBCAA and BDCAA, respectively) showed that these compounds were rapidly dechlorinated by ZVI. The bromine atoms in HAAs were removed faster than chlorine in these reactions. TBAA was observed to undergo a complete dehalogenation to form acetate while the reduction of the other examined HAAs tended to stop at the formation of MCAA. The rate of ZVI-induced dehalogenation was highest for DBCAA, in which case it exceeded the rate of BDCAA or TBAA dehalogenation by an order of magnitude. The dehalogenation of TCAA was more than two orders of magnitude slower than that of DBCAA. Comparison of the relative effects of electron and mass transfer limitations on the EC dehalogenation of HAAs by ZVI [24] showed that the dehalogenation of TBAA, DBCAA and BDCAA was limited by mass transfer. Mass transfer had a limited effect on the reduction of DBAA and BCAA while the reduction of TCAA, DCAA, BCAA and MCAA was limited by the rate of electron transfer from the cathode to the involved HAA species. The presence and concurrent EC reduction of dissolved oxygen had relatively little effect on these processes.
Higher rates of the direct EC debromination of trihalogenated HAAs were also observed in the recent study concerned with the development of EC sensors for DBPs [25]. Voltammetric EC data presented in that study indicate, in accord with the results of Hozalski et al. [23], that the EC reductive cleavage of Br from TBAA, BDCAA and DBCAA occurs in the range of potentials 0 to −0.5 V while the cleavage of Cl from these DBPs tends to become prominent in the range of potentials −0.5 to −1 V. Differences in the voltammetric data generated using a gold electrode allowed quantitating the examined HAA species at μg/L levels. In another study focused on analytical aspects of HAA quantitation [26], the reduction of MCAA, DCAA and TCAA on a catalytically active poly-Ni(II)-tetrasulfonated phthalocyanine (poly-NiTSPC) film formed on the gold substrate was compared with that on bare Au surface. The presence of the poly-NiTSPC film was observed to decrease the EC potential of the onset of the reduction of the examined HAAs by ca. 0.25 V.
2.2. Effects of EC Conditions and Electrode Materials on the EC Reduction of HAAs
The results discussed above show that the EC reduction of HAAs and other DBPs can be accelerated using electrocatalytic materials. One type of a potentially promising electrode material suitable for DBP treatment is graphene whose performance as a component of a composite 3D graphene-Cu foam electrode was examined in Mao et al. [27]. This material was used for EC reductive dehalogenation of TCAA. Voltammetric and EC impedance spectra generated for this system showed that the presence of graphene caused the reduction of TCAA to accelerate. Mechanistically, this effects was tied to the involvement of atomic H* adsorbates. The EC reduction of TCAA using this material resulted in the formation of dichloroacetic acid (DCAA) and MCAA although levels of acetic acid comparable with those of MCAA were also observed. This indicates that the use of electrocatalytic materials may allow achieving a complete dehalogenation of monohalogenated aliphatic DBPs that typically show little EC activity when conventional electrode materials are used.
Effects of electrocatalysis have been frequently examined for inexpensive materials (graphite, iron) modified with Pd, Pt or other catalysts. Li et al. [28] used a Pd/Fe-modified carbon electrode to carry out EC reduction of MAA, DCAA and TCAA at −1.5 V vs. standard calomel electrode (SCE) and pH 3. The deposition of Pd/Fe resulted in the acceleration of the degradation of these HAAs, compared with their removal using the unmodified carbon electrode. As was observed in other studies, the reductive dechlorination of TCAA and DCAA was relatively rapid but the reduction of MCAA to acetic acid was quite slow and deemed to proceed via the indirect reduction of MCAA by atomic H* generated at Pd nuclei formed on the electrode surface. The acceleration of the EC reduction of HAAs by atomic H* was also observed in another study [29] which utilized an EC-controlled bimetallic Pd/In catalyst that promoted the formation of atomic H*. The catalyst was incorporated into an EC reactor whose operations resulted in a rapid removal of TCAA yet MCAA again was observed to form as a final product.
The removal of MCAA formed in EC and related treatment processes (e.g., ZVI) may be addressed by combining the EC dehalogenation of the target with a post-treatment process. The improved performance of a combined approach was demonstrated for the degradation of TCAA by ZVI followed by the treatment of the ZVI effluent containing DCAA and MCAA by biologically active carbon [30]. This process was shown to result in a considerable reduction of MCAA concentrations.
The removal of HAAs using Pd-modified granular activated carbon (GAC) and EC-controlled Pd-modified carbon paper cathode was examined in Zhao et al. [31]. Moderate levels of the removal of HAAs at relatively low current densities (0.3 mA/cm2) were observed. For that current density, the rate of HAA removal was judged to be affected by both electron transfer and diffusion rates. For higher current densities (e.g., 0.6 mA/cm2), diffusion controls of the reaction rate became more prominent.
2.3. EC Degradation of DBPs Other Than HAAs
The EC-driven dehalogenation of DBPs other than HAAs has been addressed in a number of studies. Li et al. [32] reported data for a process that combined the sorption of DBPs by GAC and EC-driven electrolysis applied to dehalogenate the adsorbed DBPs. GAC adsorption effectively removed a representative range of DBPs (a mixture of haloacetonitriles, trihalomethanes, haloacetaldehydes, haloacetamides and chloropicrin was tested). Further treatment by electrolysis carried using the GAC cathode polarized at −1.0 V vs. standard hydrogen electrode (SHE) resulted in the degradation of the retained DBPs whose levels decreased by up to 90% for a relatively long treatment times (6 to 12 h). Activated carbons and charcoal were more effective than graphite for electrolysis, primarily due to a much higher sorption capacity of the former class of materials. The reduction of the examined DBPs was concluded to proceed via direct EC dehalogenation that results in the formation of Cl−, Br− and I− ions. The involvement in the reduction of the retained DBPs of redox-active functional groups typical for activated carbons, for instance phenols and quinones, was also hypothesized. Similar trends were reported for the case of EC dehalogenation of DBPs using a resin-impregnated graphite cathode [19]. However, the relatively low degradation efficiencies (in a 20% – 95% removal range, with most species degraded at ca. 60%) observed for > 24 h treatment duration suggested that this technology may be best applied for specific types of DBP-containing effluents.
ZVI-driven degradation of halonitromethanes (HNM) exemplified by mono-, di- and trichloronitromethans (MCNM, DCNM and TCNM, respectively) was examined in [33]. In the presence of a high ZVI dose (2 to 4 g/L of ZVI) and pH 7.5, the examined HNM species underwent a relatively slow (within one to four hours) degradation. The apparent rate of dechlorination decreased with the number of chlorine atoms in HNM molecules while the nominal end product of their dechlorination, nitromethane was observed to undergo rapid conversion by ZVI to methylamine. The degradation of TCNM and DCNM was concluded to proceed via parallel pathways of hydrogenolysis and α-elimination. These reactions take place in drinking water distribution systems containing iron material (e.g., cast or ductile iron pipe).
2.4. Specific Aspects of the Deiodination of I-DBPs
Specific aspects of the EC reduction of I-DBPs and mechanisms of deiodination were reported in Ma et al. [34]. That study examined cathodic reactions of iodoform and MIAA at varying mass transfer conditions controlled by electrode rotation and determined that these I-DBPs undergo facile deiodination at EC potentials < −0.3 V vs. standard Ag/AgCl electrode (SSCE). Mechanisms of the EC deiodination of MIAA and iodoform were shown to be similar and in both case the cleavage of iodine atoms from the parent molecule was accompanied by the formation of active iodine intermediates. This result was unexpected as the classical mechanism of EC dehalogenation favors the cleavage of X− ions in the first of the two electron transfer steps typical for reductive dehalogenation of chlorine- and bromine-containing compounds [35, 36]. Quantum chemical modeling of the electron transfer reactions involved in the reductive dehalogenation of trihalomethanes confirmed that in the case of C-Cl and C-Br bonds the formation of Cl− and Br− ions is thermodynamically preferable for the first electron transfer while the reductive cleavage of the C-I bond is likely to proceed via the formation of I. intermediate which then forms active iodine species.
The above analysis is not meant to be exhaustive but it is sufficient to demonstrate that DBP removal methods based on their EC-controlled reductive dehalogenation are fundamentally feasible, especially in the case of hybrid methods that combine EC-driven and conventional processes, for instance GAC adsorption and/or biodegradation. The EC reductive dehalogenation of DBPs has been well studied for HAAs and in some extent THMs yet for many other individual DBP species and their general classes (e.g., N- and I-DBPs, polar phenolic DBPs, etc.) more research is needed to quantify the kinetics of their EC reactions and effects of electrode properties on them. The nature of intermediates and final products formed via the EC reduction of halogenated DBPs also needs to be ascertained in more detail. Finally, practical implementation of treatment processes based on the EC reductive dehalogenation of DBPs clearly depends on further development of new types of electrocatalytic materials amenable for incorporation into EC reactors that can overcome mass transfer limitations typical for EC treatment of trace-level contaminants.
3. Electrochemical Degradation of Iodinated Contrast Media
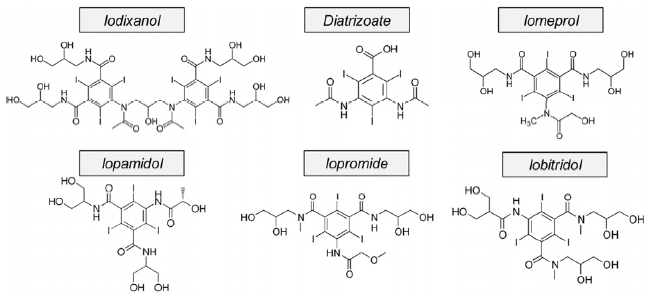
ICMs are widely encountered in hospital wastewaters, municipal wastewater and drinking water at concentrations up to 100 μg/L [12–16, 37–39]. (Chemical structures of those ICMs that have been used in prior studies of EC treatment are shown in Fig. 1). Conventional processes have little success in removing ICMs due to their high hydrophilicity and resistance to biological degradation. AOPs such as UV photolysis, sonolysis, Fenton, photo-Fenton, H2O2/UV-C, TiO2/UV-A and O3/H2O2 remove ICMs but these processes generate toxic iodinated by-products [14–16, 37–39].
3.1. Reductive Deiodination of ICMs
Reductive dehalogenation (referred to in the case of ICMs as deiodination) induced via external EC polarization of a working electrode or promoted by zero-valent metals and metal-based catalysts has shown promise in the treatment of ICMs [13, 40–42]. The susceptibility of ICMs to reductive degradation was demonstrated by Zwiener et al. [12] who examined the EC reduction of iomeprol on nickel cathode. Sequential cleavage of up to three iodine atoms from the parent molecule was observed to occur and mono- and di-deiodinated intermediates with a relatively low stability were formed in this process. The end product of the EC reduction of iomeprol was suggested to form via the transformation of the completely deiodinated iomeprol and a loss of a (C=O)CHOH group from the side chain. The authors concluded that EC degradation of ICMs may be a feasible option for the removal of these compounds from urine of patients or hospital effluents.
3.2. Reduction of ICMs by ZVI
The removal of ICMs and several antineoplastic agents by ZVI in the presence of oxygen was demonstrated in Mu et al. [43]. That study showed the removal of these species followed a pseudo-first order kinetics. The removal of iopromide was relatively slow requiring > 6 h of contact time at pH 3 to remove > 90% of the initially present iopamidol. The reaction rates were strongly affected by pH and hydrodynamic conditions which indicated the presence of mass transfer limitations.
The ZVI-driven reduction of iopamidol in the presence of monochloramine was studied in Dong et al. [41]. The contact times were as long as 60 h although the removal of iopamidol was most notable within the initial 10 h of reaction time. The degradation of iopamidol was more rapid when the corrosion of ZVI was promoted by sulfate or chloride ions at low pH (e.g., pH 5) and it became slower as the surface became passivated. The deiodination of iopamidol by increasing amounts of ZVI was accompanied by the increases of the formation of I-DBPs such as CHClI2, CHI3, MIAA, diodo- and triiodoacetic acids (DIAA and TIAA, respectively) albeit small amounts of monoiododi-chloromethane and MIAA were formed in the absence of zero-valence iron. Molar yields of iodoform and TIAA formed in the case of ZVI-induced degradation of iopamidol at pH 5 were ca. 1%. They decreased monotonically with pH and were ca. < 0.1% at pH 9. The authors expressed an opinion that given the ubiquity of ZVI in drinking water systems that about with unlined cast iron distribution pipes, the EC-controlled deiodination of iopamidol that may be present in source surface water may result in increased levels of I-DBPs.
Hu et al. [42] found that typical corrosion products found on the surface of pipes in drinking water systems (e.g., CuO and δ-MnO2) may affect the activity of peroxymonosulfate in its reactions with iopamidol. The rate of iopamidol degradation by persulfate was observed to increase in the presence of CuO or δ-MnO2, especially in the case of the former solid which was hypothesized to promote the formation of sulfate radical while CuO promoted the generation of OH radicals. The majority of the iodine released from iopamidol upon its oxidation by persulfate was oxidized to iodate yet ca. 5% of the iodine initially present in the system was transformed to active iodine species.
The degradation of iopamidol at pH 3 in the presence zero valent aluminum (ZVA) (a 1 g/L ZVA dose) was also observed to be slow. In the absence of hydrogen peroxide or persulfate only ca. 40% of the initially present iopamidol was removed within ca. 2 hours [9]. The combination of ZVA and persulftate accelerated the process in distilled water but in surface water the extent of iopamidol removal was lower, and little iopamidol was removed when wastewater was treated using ZVA/persulfate.
3.3. Effects of EC Conditions and Electrode Materials on the Degradation of ICMs
Examination of the EC degradation of 100 μM iopromide in a batch reactor equipped with a boron doped diamond (BDD) anode and a platinum cathode showed that > 90% on the initial compound could be mineralized for treatment time > 7.5 h [44]. Shorter treatment times resulted in the generation of oxidatively and reductively formed byproducts and deiodinated iopromide which was toxicologically inert. Further experiments utilizing a divided cell setup showed that the pathway of the degradation of iopromide in the examined system involved parallel reactions of reductive EC deiodination at the cathode, direct oxidation at the BDD surface and indirection radical oxidation and hydrolysis. The EC treatment of iopromide resulted in the preferential formation of iodide compared with that of iodate (90% and 10% of the total, respectively).
EC reduction of the ICM diatrizoate using three-dimensional graphite felt and graphite felt doped with nanoparticles was examined in Radjenovic et al. [13]. The presence of palladium nanoparticles resulted in a significant enhancement of the removal of diatrizoate and enabled its complete deiodination to 3,5-diacetamidobenzoic acid. When the system was employed in the treatment of hospital wastewater, diatrizoate was reduced, but the extent of EC reduction decreased as a result of competing reactions. Following the EC reduction of diatrizoate to 3,5-diacetamidobenzoic acid, the next step of EC oxidation with BDD anodes was employed to remove 3,5-diacetamidobenzoic acid. The EC reactions were employed to degrade diatrizoate in a three-compartment EC reactor operated in a continuous mode. This allowed achieving a complete deiodination of diatrizoate at a −1.7 V vs. SHE cathode potential while a predominant part (ca. 80%) of the released iodide ions was electrodialyzed into a central compartment. The oxidation of the effluent formed from the cathode compartment at the BDD anode resulted in the further removal of the reduction products.
An example of a combined system that incorporated the reductive EC dehalogenation of iopromide and the treatment of the EC effluent with a biologically enhanced anodic oxidation enhanced by acetate feeding was presented in Mu et al. [43]. The system used a granular graphite cathode whose potential was varied between −0.5 to −0.9 V vs SHE. The cathodic shift of the potential of the working electrode resulted in a strong acceleration of the EC removal of iopromide and its complete deiodination at potentials < −0.8 V although the Coulombic efficiency of the dehalogenation of iopromide at the cathode was < 1%. The Coulombic efficiency of the acetate oxidation at the graphite anode was estimated to be somewhat < 50%. Several intermediates formed upon the reduction of iopromide were observed.
The EC oxidation of six ICMs (amidotrizoate also known as diatrizoate, iopamidol, iobitridol, iodixanol, iopromide and iomeprol) in batch experiments that employed dimensionally stable anodes (DSA) resulted in the removal of up to 85% of the examined ICMs for electrolysis time exceeding 2.5 h at a constant current [45]. However, the polarity of the employed DSA electrodes was switched during the treatment, and both anodic and cathodic reactions took place in the system. The EC treatment was concluded to result in the reductive deiodination of the examined ICMs, further reduction of the resultant alkyl aromatic amides to simple amides and the de-acylation of N-aromatic amides to aromatic amines. The consumption of energy in the examined process was high (> 370 kWh/m3) yet the reported process was deemed by the authors to be viable to treat hospital wastewater or other pharmaceutical waste-contaminated streams.
The studies discussed in this section show that while the EC reductive deiodination of ICMs as well as the oxidation of their degradation products is reasonably facile, several challenges are associated with these processes. For instance, the treatment of ICM-containing solutions using zero valence metals even at their high doses tends to be slow, may require using low pHs and additional oxidants such as hydrogen peroxide or persulfate. On the other hand, the EC reduction of ICMs is facile and tends to be limited by mass transfer to the electrode surface. The EC reduction of ICMs is accompanied by the generation of unstable iodine-containing intermediates and, ultimately, completely deiodinated products such as diacetamidobenzoic acid and aromatic amines. These products have been observed to be amenable to removal by anodic oxidation and/or biological treatment.
3.4. Mechanisms of the EC Deiodination of ICMs
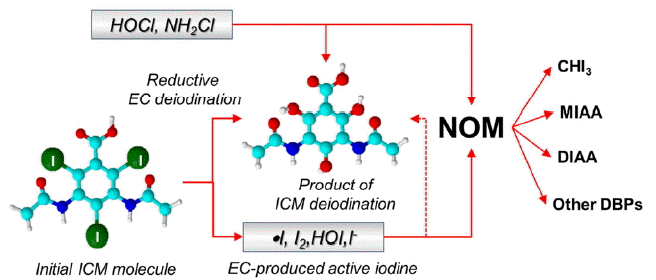
A challenging aspect of the EC treatment of ICMs is that this process is accompanied by the release of iodide (and in some extent iodate) cleaved from the parent ICMs via either EC reductive dehalogenation or in oxidative transformations of ICM molecules or intermediates formed upon their degradation. In the case of reductive EC deiodination, formation of active iodine species via the mechanisms similar to those observed for I-DBPs [34, 46] can be anticipated. Examination of the EC reduction of iopamidol and diatrizoate by the rotating ring-disk electrode (RRDE) method [46] showed unambiguously that in the range of potentials ca. −0.6 to −0.9 V vs. SSCE the EC reduction of these ICMs was mass transfer-controlled. The potentiodynamic data indicated that the EC-induced cleavage of the iodine atoms from the parent ICMs was nearly simultaneous for the three incorporated iodine atoms. The presence of natural organic matter (NOM) and model compounds such as resorcinol, catechol and guaicol did not affect the EC reduction of the ICMs but active iodine species formed as a result of the EC-induced transformations of these ICMs reacted readily with NOM and model compounds.
The formation of active iodine species in the case of the EC reduction of ICMs as well as the oxidation of the released iodide to active iodine by disinfectants can lead to the formation of I-DBPs. The scheme of reactions taking place in the case of electrochemical reduction of ICM compounds was shown in Fig. 2. Thus the control of I-DBPs formed in EC treatment of ICMs becomes a priority. This necessitates the development of reactors in which the released iodide is intercepted and/or removed, for instance by electrodialysis used in [13] or other techniques. As in the case of the EC reductive treatment of halogenated DBPs, combining EC processes with appropriate pre- and post-treatment options may be highly beneficial for the development of practically feasible EC-based treatment processes for the removal of ICMs and other TrOCs.
4. Conclusions and Suggestions for Future Research
This paper summarizes results of research concerned with the EC degradation of halogen-containing DBPs and ICMs. The efficiency of EC dehalogenation of DBPs increases with the number of halogen atoms present in a particular individual DBP species (e.g., mono-, di- and trihaloacetic acids). EC-induced reductive cleavage of bromine from parent DBPs is considerably faster than that of chlorine, and it occurs at lower cathodic potentials. EC data and quantum chemical modeling indicate that the EC reduction of iodine-containing DBPs proceed via a pathway that is different from the classical dehalogenation sequence in which the transfer of the first electron in the two-step sequential electron transfer sequence is accompanied by the cleavage of a halide X− anion. The EC reduction of I-DBPs is characterized by the formation of active iodine that reacts readily with the organic substrate thus potentially leading to the formation of secondary I-DBPs. EC oxidation of ICMs using anodes that produce reactive oxygen species (e.g., BDD, DSA) combined with the cathodic reduction of ICMs can result in a complete degradation these compounds yet iodine-containing DBPs are formed in the process. Reductive EC deiodination of ICMs using various cathode materials (e.g., nickel, platinum, graphite) is rapid and the overall rate of the EC degradation of ICM is diffusion-controlled. Deiodination of ICMs can also occur in drinking water networks via the engagement of iron surfaces and corrosion products typical for these systems. The need to intercept and remove active iodine species and iodide ions formed upon the EC degradation of ICMs is a specific challenge characteristic for the treatment of this class of TrOCs. Further progress in practically feasible EC methods to remove DBPs, ICMs and other TrOCs requires the development of more selective and stable electrocatalytic materials, elimination of mass transfer limitations via innovative design of 3D electrodes and EC reactors, and a more detailed understanding of intrinsic mechanisms of EC reactions of DBPs and TrOC at EC interfaces. Another high priority is the development of hybrid treatment systems that combine EC-controlled reactors with other pre- or post-treatment techniques.